深度揭秘“合成致死”藥物PARP抑制劑殺死癌細胞的機制_風聞
推医汇-汇聚健康行业资讯,推动学术交流。2021-05-27 12:09
“欲使其滅亡,必先使其瘋狂。”
這句話用在癌細胞身上,恐怕再適合不過了。
從本質上講,癌症就是一種基因病。當細胞內的基因突變積累到一定程度之後,細胞要麼走向衰老死亡,要麼就走向癌變。
不過這些突變在賦予癌細胞不死和無限繁殖能力的同時,也給它們的毀滅埋下了伏筆。
這個毀滅伏筆的序曲在1922年。
那一年,在哥倫比亞大學摩爾根實驗室工作的遺傳學家Calvin Bridges,在黑腹果蠅身上發現一種有趣的現象:當某兩個特定的基因同時突變失活時,會導致果蠅的死亡;而這兩個基因單獨任何一個突變失活,都不會給果蠅帶來致命的傷害。
1946年,Theodosius Dobzhansky給這種現象取了個名字,它就是今天大名鼎鼎的“合成致死”效應。
這個概念一沉寂就是51年。在考慮到癌細胞攜帶有大量基因突變之後,1997年,福瑞德·哈金森癌症研究中心的Stephen Friend敏鋭地察覺到,這個“合成致死”的理念或許可以用到癌症的治療中。
在Stephen Friend看來,正常細胞癌變是個異常的舉動。俗話説,“物極必反”,那我們乾脆就讓異常來的更瘋狂吧。
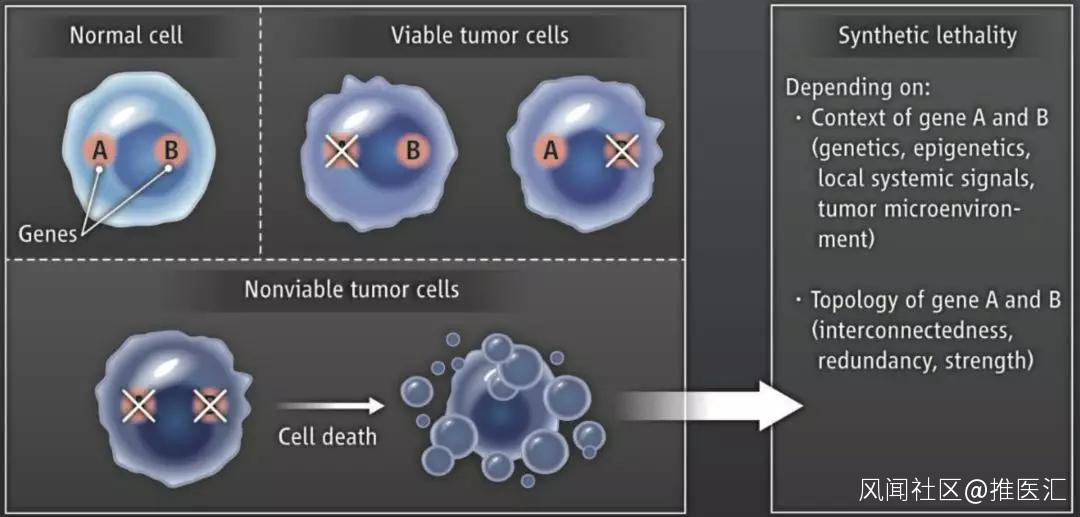
合成致死理念
這個想法很瘋狂,不過竟真的能讓癌細胞走向死亡。
2014年,全世界第一個按照“合成致死”理念設計的抗癌藥物PARP(多聚ADP核糖聚合酶)抑制劑Olaparib,獲得FDA批准用於治療卵巢癌。隨後,在2016年和2017年,PARP抑制劑Rucaparib和Niraparib先後閃亮登場。
一種全新的抗癌手段崛地而起。
“破罐子破摔”
實際上,大部分細胞從正常走向癌變,並不是説它們的基因天生就不好,而是因為在生長的過程中,細胞的DNA會不斷遭受內在和周遭各種不利因素的夾擊,例如,輻射、化學毒物、細胞自身有害代謝產物、DNA自己複製錯誤等,導致癌症相關基因發生了突變,最終導致了癌症。
據估計,人體每個細胞每天產生的單鏈DNA損傷數約為10000個,如果把其他損傷也都算上的話這個數據又要翻10倍,變成10萬個。
與DNA遭受的損傷相比,癌症的發生風險就顯得微不足道了,這主要得益於人體精密、複雜而高效的DNA修復系統。
在DNA損傷中,最嚴重的損傷是單鏈斷裂和雙鏈斷裂,不過單鏈斷裂更常見。這些斷裂如果不能得到及時、準確的修復,會使基因組變得不穩定,進而引起癌變,甚至直接導致細胞死亡。

DNA單鏈斷裂(SSB)和雙鏈斷裂(DSB)示意圖
為維持正常生理功能,細胞必須有多種DNA損傷發現和修復機制,使受損的DNA得到及時精確的修復。
對於單鏈斷裂而言,它的修復主要依賴於PARP,這個酶在人體內有17種,它們雖然長得有些像,但功能卻不盡相同。
目前的研究認為,DNA損傷修復依賴的PARPs主要包括PARP-1和PARP-2,它倆都能精準地識別DNA的傷口,並與DNA親密結合。只不過在修復DNA損傷的過程中,PARP-1發揮着90%以上的功能,PARP-2更像是個備胎。
而對於雙鏈斷裂而言,它雖然少,但是情況更嚴重,如果不能及時修復,細胞的DNA就會變得不穩定,細胞最終走向死亡。
所以雙鏈DNA斷裂有兩種主要的修復方式。一種是非同源末端連接(NHEJ)修復,它更像個緊急救火隊長,先不管修復的對不對,把斷掉的DNA連上再説。這種方法最主要的優點是快,但是非常容易出錯,一旦出大問題,對細胞來説有可能就是毀滅性的打擊。
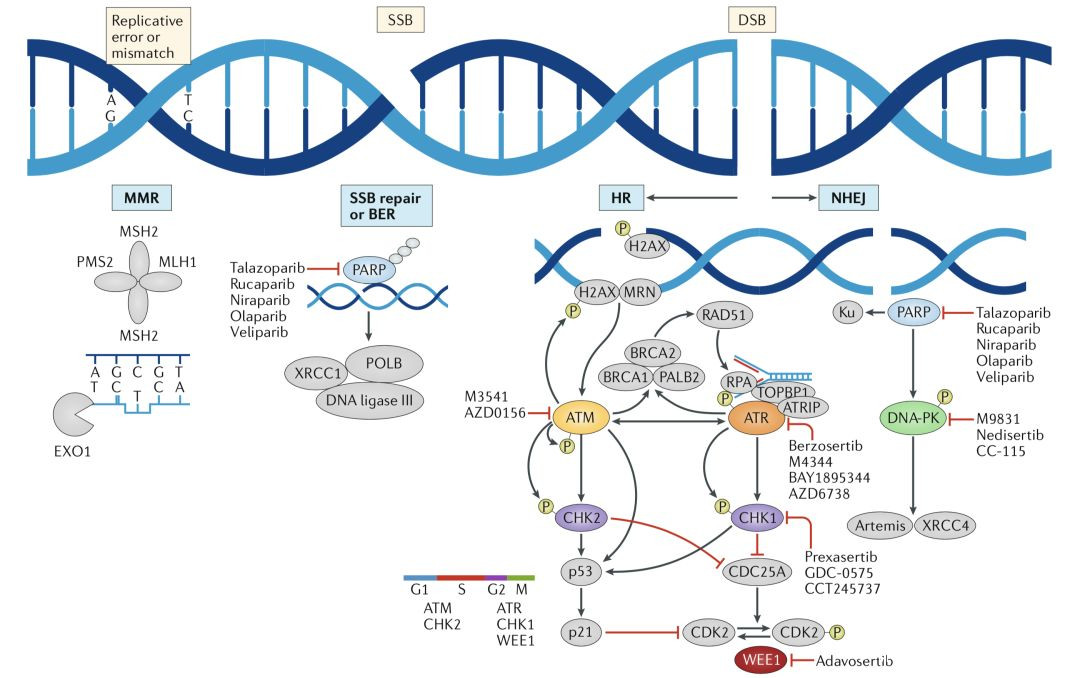
DNA斷裂的修復方式一覽
另外一種是同源重組(HR)修復途徑,參與這種修復方式的蛋白非常之多例如BRCA、ATM、RAD51等等,其中最為人所熟知的是BRCA蛋白。這種修復方式像外科手術,是一種高保真、無錯誤的修復方式。
對於癌細胞而言,既然它是基因突變導致的,那肯定是上述修復過程沒起作用,或者工作不到位造成的。
鑑於癌細胞也要維持自身基因組的穩定性,因此,作為一個“理性”的癌細胞,它們肯定不會讓上述所有的DNA損傷修復機制全部癱瘓。不過為了保持進化的活力,部分修復方式失去功能是可能的。
這也就給了科學家們可乘之機。以DNA修復為靶點,把癌細胞這個DNA已經出現大量突變的“破罐子”徹底搗毀。
2005年,“摔破”癌細胞這個“破罐子”的曙光初現。
兩個獨立研究團隊背靠背在頂級期刊《自然》發表重要研究成果,首次證實PARP抑制劑與BRCA1或BRCA2突變之間存在“合成致死”的相互作用。
合成致死治療癌症的大門打開了。
“扼住”PARP的咽喉
結合前面介紹的DNA修復機制,你會發現PARP與BRCA是一對合成致死冤家這事兒並不難理解。癌細胞的DNA再混亂,它們也還是需要維持自身基因組的穩定。
如果負責雙鏈斷裂修復的BRCA突變失活了,我們再把管單鏈斷裂的PARP抑制掉,癌細胞中每天出現的大量單鏈斷裂就會變成雙鏈斷裂,最終導致癌細胞死亡。
不過,這個合成致死的機制看似簡單,其實要設計一個優秀的PARP抑制劑並沒有那麼簡單。
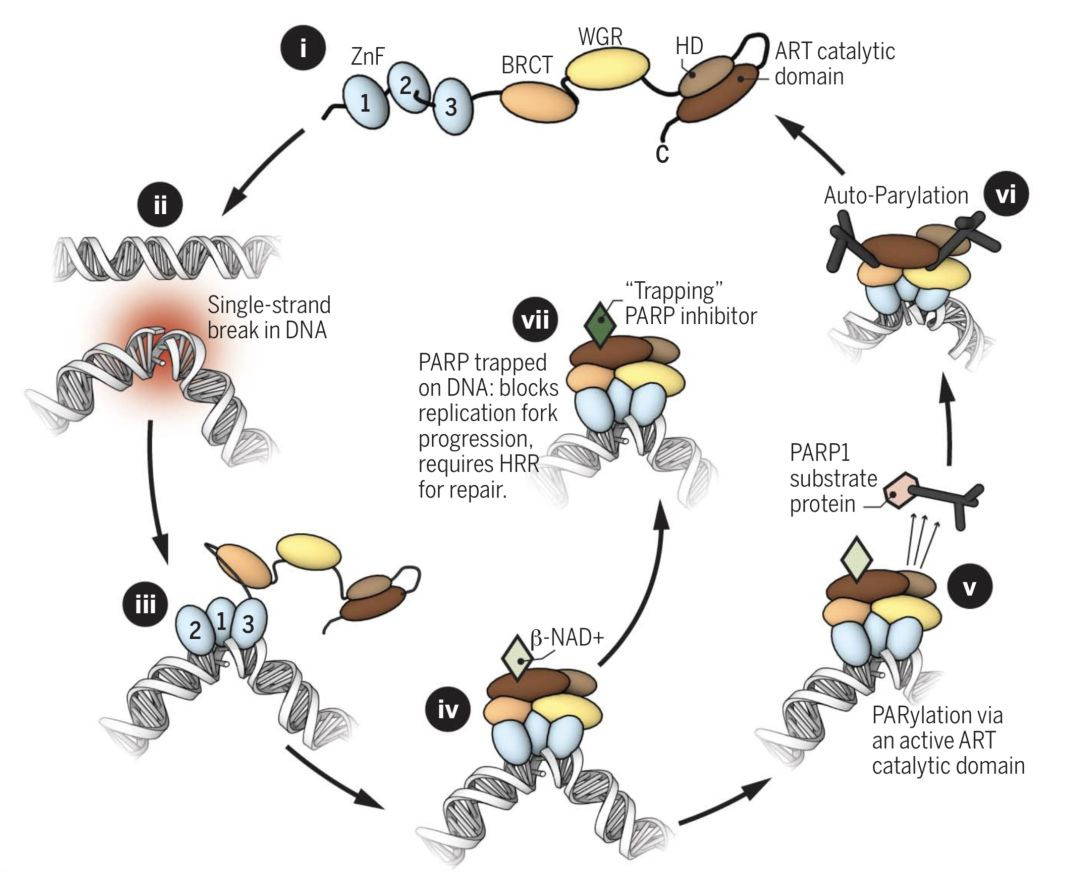
PARP抑制劑“合成致死”機制
要把這個事情説清楚,我們還得從PARP修復單鏈DNA斷裂的過程説起。
在細胞內,一旦PARP發現DNA上存在單鏈斷裂的缺口,就會立即結合上去,這種結合會激活PARP的催化活性。
此時,遊蕩在PARP周圍的煙酰胺腺嘌呤二核苷酸(NAD+,這個物質最近非常火,抗衰老、抗癌都有它的份兒)會立即與PARP的活性位點結合,結合後的複合體會把周圍參與DNA修復效應子統統拉過來,填補上DNA斷開的缺口。於此同時,染色質也會變得鬆弛,PARP複合體就順利從損傷缺口脱離下來,回到之前的失活狀態待命。
在這個修復的過程中,NAD+與PARP的結合,就是那個關鍵的點。
實際上,早在30年前,小分子煙酰胺類似物就被證明可以競爭性抑制這個過程,並增強DNA損傷劑硫酸二甲酯的細胞毒性。
目前在臨牀中使用的所有PARP抑制劑,都有一個與NAD+競爭結合PARP的煙酰胺部分,因此它們抑制PARP催化活性的能力是類似的;然而,由於不同的抑制劑結構存在較大差異,它們對不同PARP家族成員的選擇性存在一定的差異。
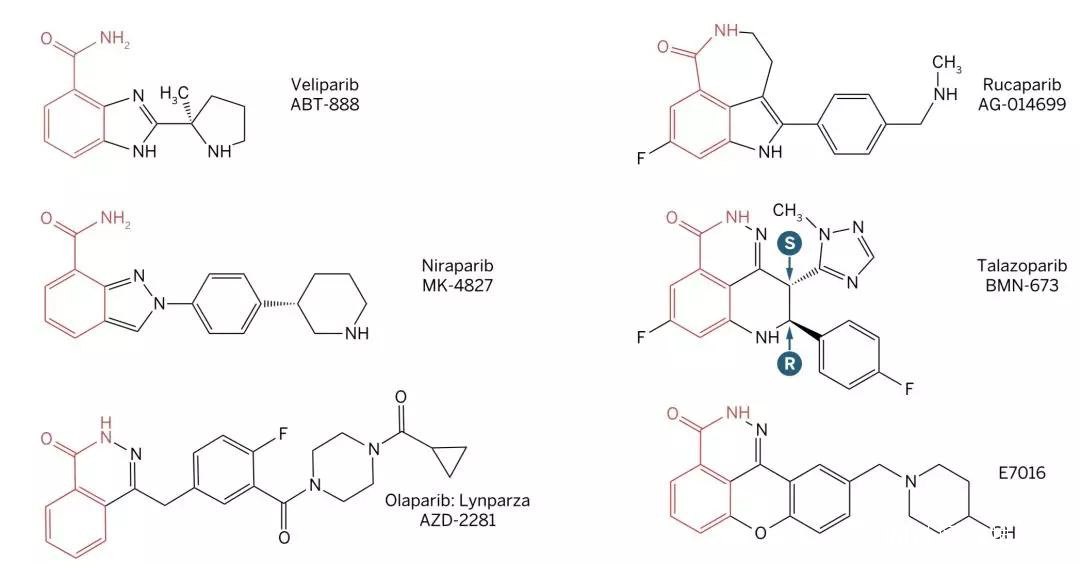
PARP抑制劑結構比拼:紅色部分為共通部分
可別小看了這個差異,畢竟科學家對PARP-3的認知還不夠。
雖然從結構上看,PARP-3與PARP-1也長得很像,但PARP-3在組織分佈、生物學功能方面,與PARP-1卻表現出很大的不同[。
除此之外,之前還有研究表明,當PARP-1表達被抑制之後,PARP-2的表達會代償性地增加,以頂替PARP-1的職能;但PARP-3卻不會在PARP-1和PARP-2表達被抑制後代償性增加。而且也有研究表明,特異性抑制PARP-1和PARP-2,但不抑制PARP-3,就能讓腫瘤消退。
這些似乎都表明PARP-3有其獨特的生物學功能。
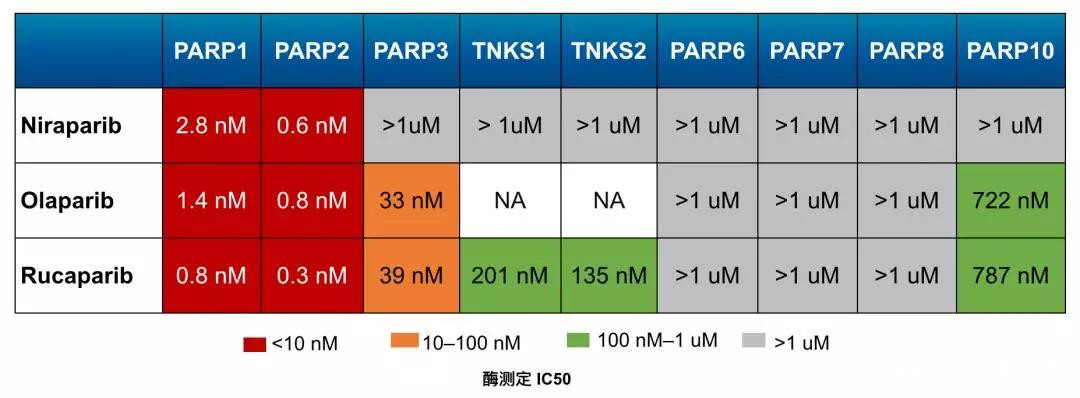
部分PARP抑制劑選擇性的比較
由此可見,PARP抑制劑抑制PARP-3的活性,可能不僅沒有抗癌效果,而且可能還有意想不到的副作用。
“誘捕”PARP
在研究PARP抑制劑的過程中,科學家們還發現了一個很奇怪的現象。
PARP抑制劑對癌細胞的殺傷力大於敲除PARP基因本身,這意味着PARP抑制劑的抗癌效果不僅僅在於抑制PARP的活性,背後可能還有其他的原因。
後來科學家發現,這個現象要歸結於PARP抑制劑對PARP的“誘捕”作用。
所謂“誘捕”作用,説的是PARP抑制劑競爭性結合到PARP酶上之後,會導致與受損DNA結合的PARP-1和PARP-2被困在DNA上下不來了,同時直接造成其他的DNA修復蛋白也結合不上來了。後果是,DNA斷裂不僅不能被修復,而且還從單鏈斷裂變成雙鏈斷裂,最終導致細胞死亡。
實際上,科學家已經認識到,“誘捕”PARP並把它“釘”在DNA上,才是PARP抑制劑消滅癌細胞的最大殺器。因此,在比較單一PARP抑制劑抗癌活性時,必須基於其捕獲效力。
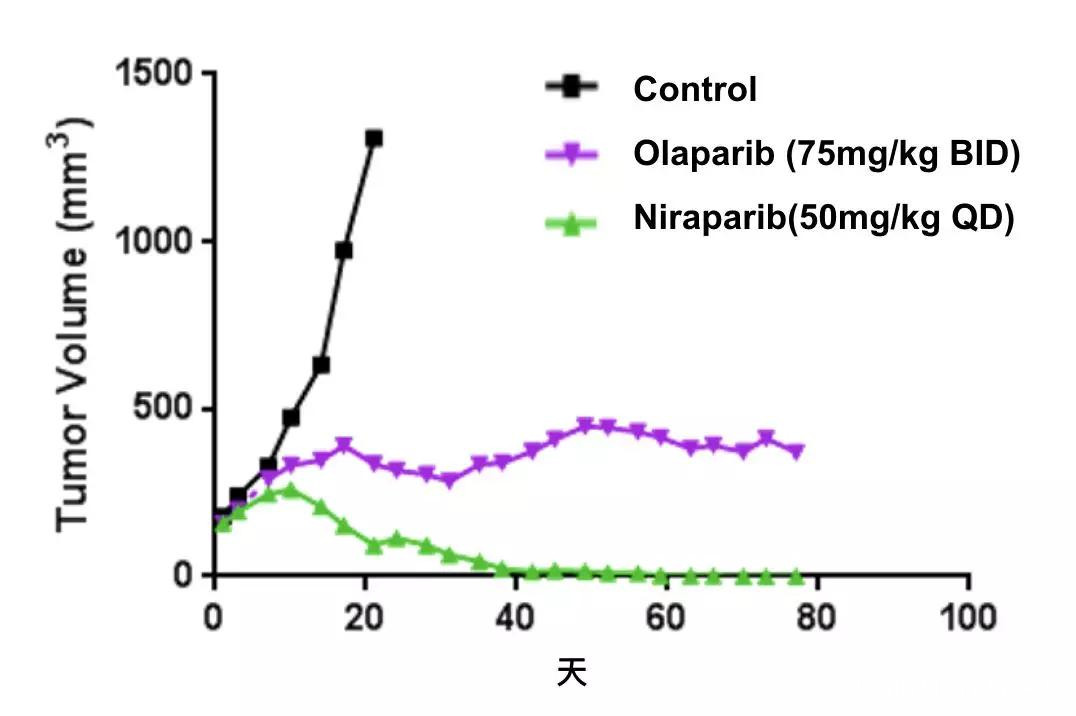
PARP抑制劑在BRCA2突變模式小鼠中的效果
它們捕獲能力之間的這種差異,也反映在了抗癌效果上。
與眾不同,潛力無限
可能你已經發現了,上面介紹的都是PARP抑制劑與BRCA突變之間的協同致死作用,但是以BRCA為代表的同源重組通路並非只有BRCA這一條路,對於BRCA基因沒有突變的癌細胞,PARP抑制劑是不是也有效果呢?
其實,2005年的研究就已經表明:PARP抑制劑對於BRCA沒有突變的癌細胞也有殺傷力。
只不過與攜帶BRCA突變的癌細胞相比,BRCA沒有突變的癌細胞對PARP抑制劑的敏感性差了近1000倍。
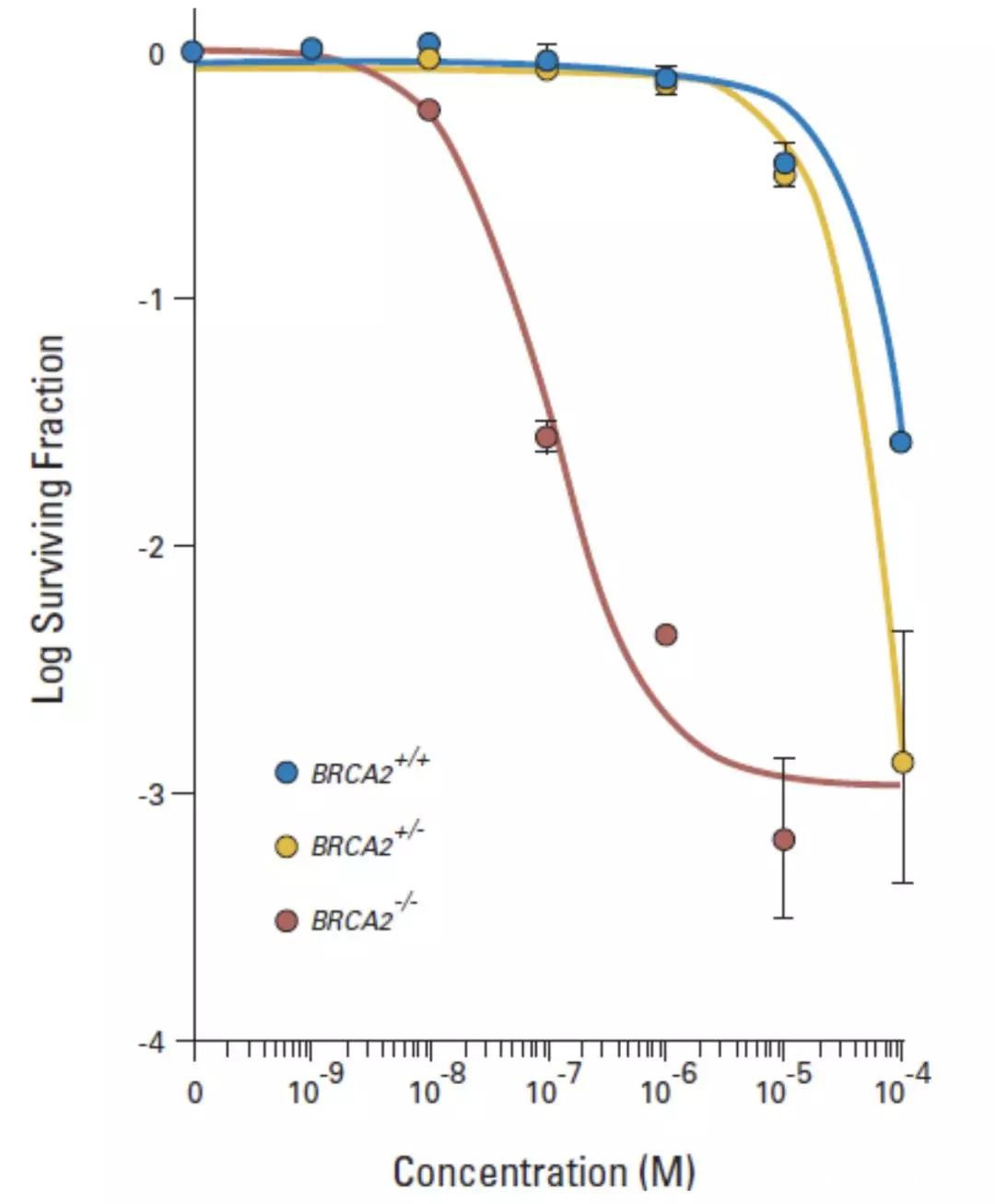
BRCA突變比不突變對PARP抑制劑更敏感
這也就意味着,對於那些BRCA基因沒有突變的腫瘤,在使用PARP抑制劑治療時,需要更高的藥物暴露,才能到達與BRCA突變的腫瘤同樣的效果。
當然,我們對於PARP抑制劑的認知還剛剛起步,它應該還有很多未知的技能等着我們去發現。
例如德州大學MD安德森癌症中心的研究團隊發現,抑制腫瘤細胞的PARP修復通路,竟然可以觸發STING免疫通路,進而募集殺傷性T細胞進入腫瘤。
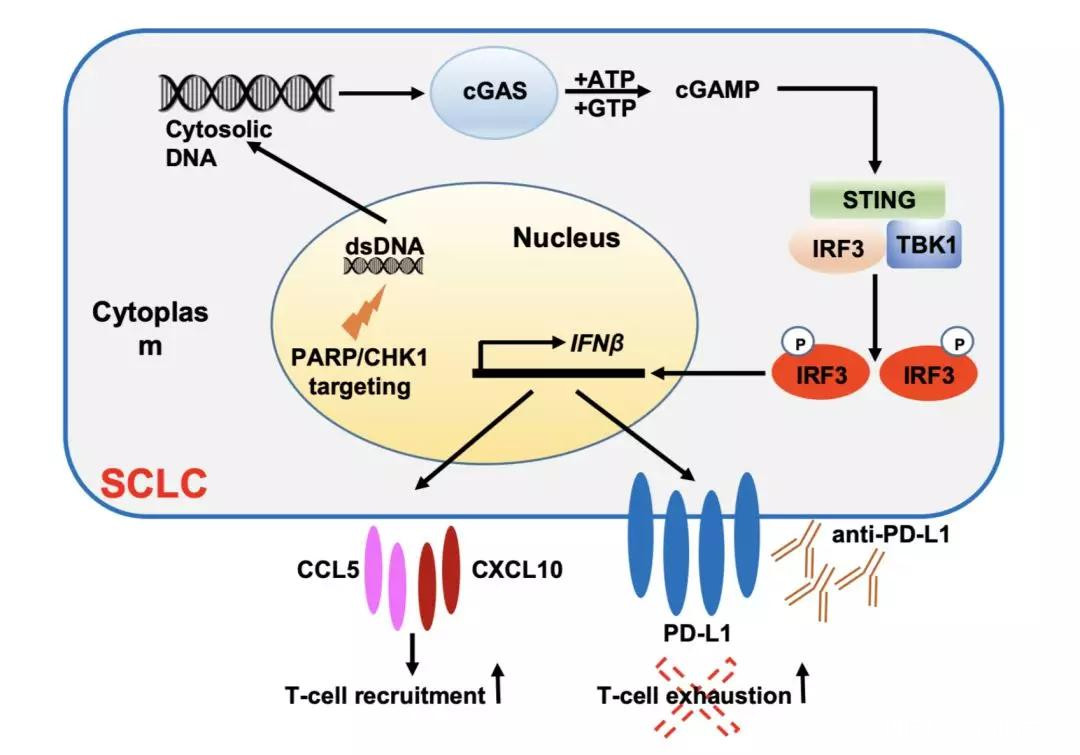
抑制PARP激活免疫通路的機制
這個研究暗示,PARP抑制劑聯合免疫檢查點抑制劑將大有可為。