單分子全長轉錄本異構體測序揭示了心肌細胞中與疾病相關的 RNA 異構體_風聞
观察员2号-2021-07-12 09:21
抽象的
選擇性剪接產生不同的 RNA 異構體,控制真核生物的表型複雜性。它的故障是許多疾病的基礎,包括癌症和心血管疾病。在全基因組範圍內對 RNA 同種型進行比較分析一直很困難。在這裏,我們建立了一個實驗和計算管道,該管道執行從頭轉錄本註釋並準確量化來自 cDNA 序列的轉錄本異構體,全長異構體檢測準確度為 97.6%。我們生成了一個可搜索的、定量的人類轉錄組註釋,其中包含 31,025 個已知和 5,740 個新的轉錄亞型 ( http://steinmetzlab.embl.de/iBrowser/ )。通過在 RNA 結合基序蛋白 20 ( RBM20) 與侵襲性擴張型心肌病 (DCM) 相關的突變,我們確定了 107 個心臟基因中的 121 個差異表達的轉錄亞型。我們的方法能夠對複雜的轉錄結構進行定量剖析,而不僅僅是識別單個外顯子的包含或排除,如發現由 RBM20 突變錯誤剪接的IMMT同種型為例。因此,我們實現了獨立於現有轉錄亞型註釋的直接差異表達測試的途徑,提供更直接的生物學解釋和更高分辨率的轉錄組比較。
介紹
幾乎所有的人類多外顯子基因都被交替剪接1 , 2 , 3 , 4,允許單個基因產生多個 RNA 異構體,從而產生不同的蛋白質異構體5 , 6,從而驅動真核生物1 的表型複雜性。許多疾病,包括癌症、神經系統疾病和心血管疾病,都與選擇性剪接失調有關,例如心臟特異性選擇性剪接調節因子RBM20 的突變,導致擴張型心肌病 (DCM) 7 , 8 , 9. 研究選擇性剪接的常規方法是短讀長 RNA-seq 1 , 4 , 10 , 11 , 12 , 13,它對 RNA 片段進行測序,因此只能檢測整個基因或單個外顯子的表達變化。儘管 Kallisto 14或 Salmon 15等軟件工具允許使用短讀長 RNA-seq 進行轉錄水平定量,這需要參考註釋或從頭轉錄組組裝。後者是發現新同種型所需的,從短讀數據中尤其具有挑戰性。因此,短讀長 RNA-seq 對於定量檢測單個 RNA 異構體並不理想。包括Pacific Biosciences(PacBio)和Oxford Nanopore Technologies(ONT)在內的長讀長測序技術的出現,為更全面地分析可變剪接提供了一種新工具。PacBio 的一項初始全基因組研究使用了 476,000 個讀數16,這使得從頭轉錄物識別成為可能,但不足以進行準確的量化。因此,後續的用PacBio或ONT研究集中於使用富集方法量化所選擇的基因,缺少基因組範圍的型材17,18,19,20。最近的研究已經使用長讀測序小鼠量化轉錄物21,22和乳腺癌細胞23,然而樣品之間沒有定量差異進行了評估。此外,長讀長測序剛剛開始在單細胞轉錄組學中用於研究轉錄異質性24 , 25. 應用長讀長 RNA-seq 來確定轉錄本的全局表達變化是具有挑戰性的,因為:(1) 低測序深度通常不允許足夠的基因組覆蓋進行準確定量;(2) 高測序錯誤率需要新的計算方法來實現轉錄本量化、分類和可視化;(3) 由於測序偽影,例如逆轉錄酶 (RT) 的不希望的模板轉換和寡核苷酸 (dT) 21 的脱靶引發,已識別的未註釋轉錄本的錯誤發現率很高混淆下游分析。因此,迄今為止,還不可能進行定量、全基因組差異表達分析來大規模檢測同種型變化。這使得很難確定許多涉及剪接的疾病的分子機制。在這裏,我們證明了單分子全長 RNA 測序能夠識別疾病相關的轉錄亞型。
結果和討論使用 ONT 測序生成長讀長 RNA-seq 數據集
我們使用 cDNA ONT 測序為人誘導多能幹細胞衍生的心肌細胞 (iPSC-CM) 生成了一個大型長讀長數據集,並開發了一個工作流程,該工作流程可準確定量地測量和比較基因組範圍內的全長剪接異構體,而不依賴於現有註釋。來自具有和不具有 DCM 相關RBM20突變的人類 iPSC-CM(每個具有兩個獨立克隆)的 RNA 以及摻入對照(ERCC 26和亮片27),被轉化為全長 cDNA 並使用 ONT MinION 技術進行測序(方法部分、補充圖 1和補充表 1)。我們總共生成了 2100 萬個高質量讀數(平均 qscore ≥6,方法部分,補充圖 2),並量化了總共 11,707 個基因的 36,765 個轉錄亞型,覆蓋了蛋白質編碼的 53.8%(10,682/19,847)基因在人類基因組(圖 1,補充圖 3,和補充表格 1 - 3)。相比之下,我們具有約 12 億對末端讀數的互補短讀數據識別了 16,726 個蛋白質編碼基因(讀數 >1)。
圖 1:全長人類 iPSC-CM 轉錄組的複雜圖景。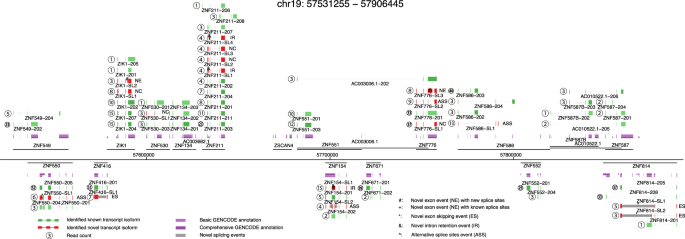 以 19 號染色體區域 (chr19:57531255-57906445) 為例,對人類 iPSC-CM 中全長剪接亞型的全基因組測量。基因位點表示為水平黑線,分別在基因組軸上方或下方的 + 或 – 鏈上帶有註釋基因。摺疊 GENCODE 全面和基本的註釋以深紫色和紫色軌道呈現。已知的轉錄異構體顯示為綠色軌跡,以前未識別的轉錄異構體顯示為紅色。已知轉錄本異構體未通過轉錄本過濾標準以淺綠色顯示。每個識別出的轉錄本的閲讀計數以圓圈中的數字給出。基於新的剪接事件(用灰色框和文本符號表示的位置),新的轉錄本異構體被分類為新的外顯子組合(NC)、新的外顯子(NE)、新的內含子保留 (IR)、新的外顯子跳躍 (ES) 或新的替代剪接位點 (ASS)。不顯示通過成績單閲讀的可疑小説。
以 19 號染色體區域 (chr19:57531255-57906445) 為例,對人類 iPSC-CM 中全長剪接亞型的全基因組測量。基因位點表示為水平黑線,分別在基因組軸上方或下方的 + 或 – 鏈上帶有註釋基因。摺疊 GENCODE 全面和基本的註釋以深紫色和紫色軌道呈現。已知的轉錄異構體顯示為綠色軌跡,以前未識別的轉錄異構體顯示為紅色。已知轉錄本異構體未通過轉錄本過濾標準以淺綠色顯示。每個識別出的轉錄本的閲讀計數以圓圈中的數字給出。基於新的剪接事件(用灰色框和文本符號表示的位置),新的轉錄本異構體被分類為新的外顯子組合(NC)、新的外顯子(NE)、新的內含子保留 (IR)、新的外顯子跳躍 (ES) 或新的替代剪接位點 (ASS)。不顯示通過成績單閲讀的可疑小説。
全尺寸圖片開發 FulQuant 以定義全長轉錄組當前使用長讀長測序定量同種型的方法的主要問題是測序偽影和導致高錯誤轉錄發現的高測序錯誤。為了應對這些挑戰,我們建立了全長轉錄本量化 (FulQuant) 的計算方法,該方法集成了 ONT cDNA 序列的準確全長讀取識別、轉錄本量化和可視化。這種新方法允許從頭轉錄本註釋,並採用嚴格而複雜的標準過濾讀數、比對和轉錄本,以排除 RT 和測序錯誤中的偽影,並根據其剪接位點將讀數分組到轉錄本中,生成一組高度可信的具有丰度估計的轉錄本(方法部分和補充圖 4)。為了評估 FulQuant 識別同種型的準確性,我們確定了其在合成測序摻入序列(亮片,摻入佔總 RNA 的 3%)上的性能,其濃度範圍約為 10 6倍,涵蓋了觀察到的基因表達的動態範圍跨越人類轉錄組27. 這些spike-ins 代表72 個人工基因位點,具有多個編碼156 種替代異構體的外顯子,以模擬不同序列、結構、丰度和長度的人類剪接異構體。我們鑑定了 156 種亮片同種型中的 82 種,包括 50% 最集中的同種型中的 90% (70),佔所有亮片 RNA 分子的 99.95%。我們的同種型鑑定真陽性率,定義為鑑定的參考轉錄物與所有報告的轉錄物的比率,基於所有亮片同種型為 97.6%,最大的亮片同種型為~7 kb(圖 2a)。總之,人類 GENCODE 註釋中所有剪接轉錄本的 98.1% 低於這個 7 kb 閾值(圖 2b)。對於短於 1728 bp 的同種型,真陽性率為 100%,其長度大於所有人類剪接轉錄本的 70%。FulQuant 的識別準確度高於現有的長讀取管道 FLAIR 28,當使用默認設置(補充文本)對我們的亮片數據集進行基準測試時,它產生了 30% 的真陽性率。FulQuant 在轉錄本識別方面的改進主要是由於去除了合成對照中出現的 RT 和測序中的人工製品造成的假陽性轉錄本。使用 FulQuant,亮片的量化與輸入濃度呈線性相關(平均 Pearson 相關係數 0.85,補充圖 5)。此外,98.8% 的轉錄本邊界位於帶註釋的轉錄本的 20 bp 內(補充圖 6),表明可以可靠地捕獲全長轉錄本,而不依賴於註釋。同樣,當我們將 FulQuant 應用於我們的人類 iPSC-CM 數據集時,我們觀察到技術和生物學重複之間的高度相關性(補充圖 7),以及全長轉錄本的準確邊界定義(補充表 2和3)。FulQuant 的量化在生物複製之間產生了比 FLAIR 更好的一致性(補充圖 7),這可能是由於假陽性轉錄本的數量減少了。
圖 2:FulQuant 方法準確識別 iPSC-CM 中的轉錄異構體。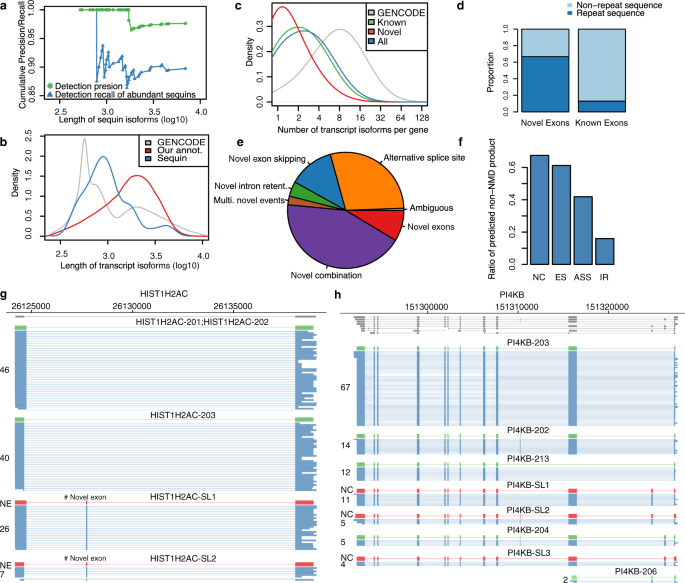 a FulQuant 方法的長度相關精度(綠色)和召回率(藍色),使用全部或最豐富的 50% 合成亮片對照進行估計。b GENCODE 中轉錄本、我們鑑定的同種型和亮片對照的長度分佈。a、b與相同的 x 軸垂直對齊,以説明亮片對人類轉錄本估計的準確性的相關性。c基於 GENCODE 綜合註釋和本研究中鑑定的轉錄本(分為已知、新穎和全部),每個基因的同種型數量分佈。d包含重複序列的已知外顯子的比例與已識別的新外顯子的比例相比。電子按新剪接事件類型劃分的新轉錄本異構體的百分比:新外顯子 (NE)、新內含子保留 (IR)、新替代剪接位點 (ASS)、新外顯子跳躍 (ES) 和已知外顯子的未註釋組合 (NC) . 模稜兩可的成績單是那些有對齊問題的成績單。f每個新轉錄物類別中預測的非 NMD 產品的比率。g , h分別具有已知外顯子和新外顯子未註釋組合的基因示例。綜合 GENCODE 註釋中的已知同種型以灰色軌跡顯示。未顯示具有 <5 個讀數的同種型。源數據作為源數據文件提供。
a FulQuant 方法的長度相關精度(綠色)和召回率(藍色),使用全部或最豐富的 50% 合成亮片對照進行估計。b GENCODE 中轉錄本、我們鑑定的同種型和亮片對照的長度分佈。a、b與相同的 x 軸垂直對齊,以説明亮片對人類轉錄本估計的準確性的相關性。c基於 GENCODE 綜合註釋和本研究中鑑定的轉錄本(分為已知、新穎和全部),每個基因的同種型數量分佈。d包含重複序列的已知外顯子的比例與已識別的新外顯子的比例相比。電子按新剪接事件類型劃分的新轉錄本異構體的百分比:新外顯子 (NE)、新內含子保留 (IR)、新替代剪接位點 (ASS)、新外顯子跳躍 (ES) 和已知外顯子的未註釋組合 (NC) . 模稜兩可的成績單是那些有對齊問題的成績單。f每個新轉錄物類別中預測的非 NMD 產品的比率。g , h分別具有已知外顯子和新外顯子未註釋組合的基因示例。綜合 GENCODE 註釋中的已知同種型以灰色軌跡顯示。未顯示具有 <5 個讀數的同種型。源數據作為源數據文件提供。
全尺寸圖片人類 iPSC-CM 全長轉錄組的全局分析我們的數據揭示了已知和新型轉錄本亞型的全基因組複雜可變剪接模式。通過將我們的轉錄異構體與人類 GENCODE 註釋 (V24) 進行比較,我們確定了 36,765 個轉錄異構體,其中 5740 個(15.6%)是新的。例如,我們鑑定了鋅指基因ZNF211 的八個已知和四個新轉錄本(圖 1)。與綜合註釋集 CHESS (v2.2) 29 相比,3028 (8.2%) 是新穎的(補充文本和圖 8)。我們的全基因組全長同種型數據集可通過可搜索瀏覽器獲得,該瀏覽器是我們開發並免費提供的 ( http://steinmetzlab.embl.de/iBrowser/ )。
我們鑑定的轉錄本異構體的長度從 177 bp 到 10,718 bp,中位數為 1768 bp(圖 2b和補充圖 9)。長度分佈類似於 GENCODE 全長蛋白質編碼和 lincRNA 轉錄本(補充圖 10),但是我們沒有捕獲大的轉錄本,例如TTN同種型(大小約為 100 kb,心肌的主要結構成分)可能是由於 cDNA 合成的 RT 限制。確定了每個基因的兩個可變剪接轉錄本同種型的中位數(圖 2c)。值得注意的是,通過將我們的方法僅應用於一種細胞類型,即 iPSC-CM,我們已經鑑定了 24.1% 的蛋白質編碼同種型(n = 80,901) 涵蓋 GENCODE 綜合註釋中的 9946 個基因,該註釋累積了來自各種人類細胞系、組織和羣體的數據。這反映了我們的長讀長方法的綜合性質和深度,並表明人類基因組中所有剪接異構體的數量和複雜性可能被低估了。
異常剪接可能會產生新的轉錄本,可用於診斷和藥物開發。值得注意的是,在我們的數據集中檢測到的新外顯子富含由 RepeatMasker 定義的重複序列(圖 2d),其中 21% 的新外顯子與 SINE/Alu 序列重疊。除了更高程度的內含子保留(IR,4.1%)外,大多數新型轉錄本異構體還包含替代剪接位點(ASS,29.2%)、新型外顯子跳躍(ES,12.7%)、新型外顯子(NE,8.6%)、和未註釋的已知外顯子組合(NC,42.9%,圖 2e,g,h)。為了評估我們管道的性能,我們使用 RT-PCR 的獨立片段分析成功驗證了 33 個新剪接事件(外顯子跳躍、內含子保留和新外顯子)中的 26 個(方法部分,補充圖)。 11和12以及補充表 6)。在蛋白質編碼潛力方面,我們確定的新型轉錄本異構體平均每四個基因添加 1 個潛在的蛋白質編碼異構體(圖 2f和補充文本)。支持我們的數據的分辨率,我們還確定從相鄰基因(補充圖45個潛在剪接多順反子轉錄物 13被從線粒體基因組(補充圖轉錄)和未剪接轉錄多順反子 14)30,31。
人類 iPSC-CM 中協調外顯子使用事件的分析外顯子的協調錶達是許多調節機制的基礎23。它的分析也提供了重要的見解選擇性剪接的策略32,33。特別是在心臟中,TPM1、2和 3等基因中的互斥外顯子對已被用作模型系統來研究剪接調節34。我們的長讀長數據首次系統地分析了心臟細胞中整個轉錄本亞型的相鄰和遠距離外顯子之間的相互依賴性。我們在 722 個基因中鑑定了 2793 個顯着共調控的外顯子對(調整後的P < 0.001,相互排斥和包含關聯,方法部分和補充表 4)。例如,我們在心臟特異性基因TPM1 中檢測到有據可查的外顯子對關聯,該基因包含相鄰的互斥外顯子 2 和 3,它們代表平滑肌和骨骼肌35之間的肌肉類型轉換。此外,我們在TPM1轉錄本亞型中鑑定了 6 個進一步的共關聯事件,包括 4 個遠端對,例如外顯子 2 和 11 之間的相互包含對和外顯子 7 和 11 之間的互斥對(圖 3a)。另一個突出的例子是MYL7(圖 3b),一個心臟功能的關鍵基因,包含 18 個相互包含 (6) 和排斥 (12) 的外顯子對。該基因中所有測試的外顯子對 (25) 中超過 70% 顯着相關,並且大多數 (61%) 位於遠端,表明該基因的剪接調控複雜。總的來説,我們觀察到一個或兩個外顯子內的相鄰共關聯比遠端共關聯更常見(補充圖 15)。由於新生 RNA 的剪接以極快的速度共轉錄發生(“50% 的剪接在 3’ 剪接位點合成後約1.4 秒內完成”)36,我們推斷這在理論上有利於相鄰的新合成的快速相關剪接外顯子。
圖 3:人類 iPSC-CM 中的外顯子相關事件。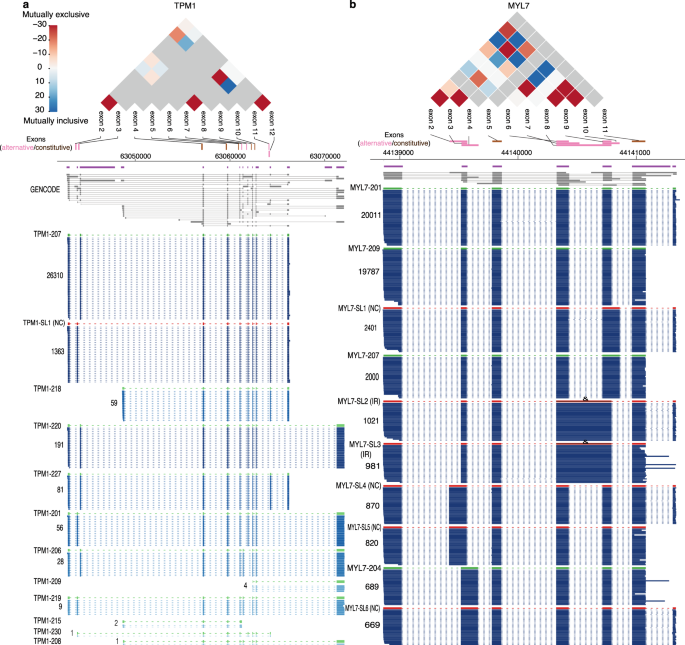 TPM1 ( a ) 和MLY7 ( b ) 顯示為包含多個相鄰和遠端相互包含或排斥的替代外顯子的基因的例子。替代外顯子對之間的共關聯強度(p值的log10 )在互斥(紅色,-)和包含對(藍色,+)的三角形圖中説明,而灰色表示未測試的外顯子對。紫色和灰色註釋分別用於摺疊和展開的 GENCODE 綜合註釋。藍色讀數與圖1 的對數刻度相同 暗度與讀取計數相關。僅顯示了用於我們的共關聯分析的外顯子。為清楚起見,未顯示低丰度的轉錄本異構體。源數據作為源數據文件提供。
TPM1 ( a ) 和MLY7 ( b ) 顯示為包含多個相鄰和遠端相互包含或排斥的替代外顯子的基因的例子。替代外顯子對之間的共關聯強度(p值的log10 )在互斥(紅色,-)和包含對(藍色,+)的三角形圖中説明,而灰色表示未測試的外顯子對。紫色和灰色註釋分別用於摺疊和展開的 GENCODE 綜合註釋。藍色讀數與圖1 的對數刻度相同 暗度與讀取計數相關。僅顯示了用於我們的共關聯分析的外顯子。為清楚起見,未顯示低丰度的轉錄本異構體。源數據作為源數據文件提供。
全尺寸圖片鑑定RBM20突變體中錯誤拼接的全長轉錄本亞型異常剪接與多種疾病有關,但確定致病事件具有挑戰性。我們使用全長轉錄本測序的方法為研究不同狀態之間的差異異構體表達提供了機會,這將使健康和疾病之間的直接比較成為可能。要制定一個量化的方式來確定樣品類型之間的差異表達的轉錄亞型,我們應用我們的管道研究全長剪接異構體是如何通過在心臟特異性剪接突變的調節影響RBM2037,38,39。20元突變(例如 R634Q 在其富含精氨酸/絲氨酸的結構域內)導致 DCM 的侵襲性形式,並且短讀長測序表明RBM20靶外顯子在大鼠37、小鼠40、豬41和人類心臟細胞中被錯誤剪接38 , 42. 然而,在這些目標中的許多目標中,尚不清楚這些差異剪接的外顯子屬於哪種異常剪接的轉錄亞型,以及是否可能會或可能不會產生故障蛋白質,或者可能只是導致正確剪接蛋白質的缺失。除了-6 M讀取為R634Q突變體,我們還產生9M讀取的P633L突變體,一個新的致病突變(脯氨酸到亮氨酸改變在氨基酸位置633),我們最近發現42,43. 我們使用 R634Q 和野生型數據進行了從頭轉錄本註釋。雖然這種方法不能識別僅在 P633L 突變體中看到的新同種型,但它使我們能夠定量比較所有突變體中在 R634Q 和野生型中檢測到的同種型的表達水平。為了將攜帶突變體 R634Q 和 P633L 的 iPSC-CM 的全長轉錄組與野生型RBM20進行比較,我們使用 DESeq2 44對量化的轉錄本進行了差異異構體表達分析(補充表 3)。我們在 107 個差異表達的基因中鑑定了 121 個轉錄亞型(調整後的P < 0.001,圖 4a,補充表 5,和方法部分)。兩種突變體都顯示出相似的表達水平(相關係數 = 0.77),並且在使用差異表達分析相互比較時沒有顯着差異(補充圖 16)。通過使用 Kallisto(方法部分、補充文本和補充圖17和18)對短讀長數據進行相同的分析運行,並使用 Kallisto 進行轉錄同種型定量,我們對通過長讀長測序鑑定的超過 80% 的候選進行了概括 。對這 107 個基因的基因本體分析顯示心臟功能富集,如橫紋肌發育、心臟收縮調節、離子轉運和基於肌動蛋白絲的過程(調整後的P < 0.05),支持了心臟功能中的RBM20(補充文字和補充圖 19)。
圖 4:RBM20突變體 R643Q 和 P633L 中差異表達的全長轉錄本異構體。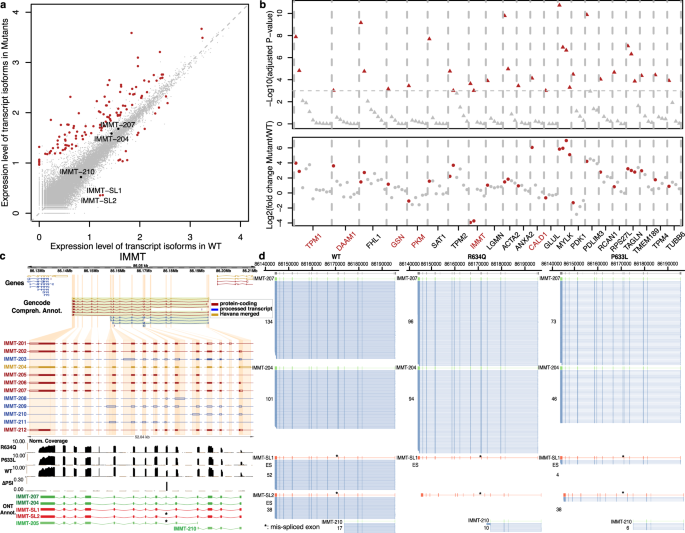 WT 和突變體中平均全長同種型表達水平的散點圖。每個點代表一個異構體,紅點突出顯示差異表達的異構體(調整後的P < 0.001)。對於異構體IMMT基因標記。b含有差異表達同種型的基因。頂部,每個同種型(三角形)的差異表達分析的顯着性水平,顯着性為紅色。底部,每個同種型(點)的表達水平的倍數變化,紅色顯着。紅色的基因名稱是基因差異表達僅佔其同種型的比例,在總基因表達水平上沒有顯着變化。僅顯示具有三個以上表達同種型的基因。ç IMMT基因與 GENCODE 註釋,短讀數據和本研究的長讀數據中鑑定的轉錄亞型。從上到下的軌跡:IMMT基因座周圍 GENCODE 轉錄本的摺疊視圖、IMMT GENCODE 轉錄本的擴展視圖、突變體和 WT 的短讀覆蓋、短讀數據中推斷的突變體和 WT 之間的 delta-PSI,以及已知(綠色)和在長讀數據中識別的新(紅色)轉錄本。GENCODE 轉錄本類型用紅色(蛋白質編碼)、藍色(處理過的)和金色(哈瓦那合併)進行顏色編碼。編碼區域由 GENCODE 註釋軌跡中的填充顏色突出顯示。d RBM20中IMMT同種型表達的並排比較R634Q、P633L 突變體和 WT。兩種新的紅色轉錄本同工型IMMT-SL1和IMMT-SL2僅在 WT 樣品中表達。已知的 GENCODE 轉錄本為綠色,原始長讀為藍色。錯誤拼接的外顯子 6 用星號表示。IMMT-205和IMMT-208由於讀取計數低(所有樣本中為 1)而被省略。源數據作為源數據文件提供。
WT 和突變體中平均全長同種型表達水平的散點圖。每個點代表一個異構體,紅點突出顯示差異表達的異構體(調整後的P < 0.001)。對於異構體IMMT基因標記。b含有差異表達同種型的基因。頂部,每個同種型(三角形)的差異表達分析的顯着性水平,顯着性為紅色。底部,每個同種型(點)的表達水平的倍數變化,紅色顯着。紅色的基因名稱是基因差異表達僅佔其同種型的比例,在總基因表達水平上沒有顯着變化。僅顯示具有三個以上表達同種型的基因。ç IMMT基因與 GENCODE 註釋,短讀數據和本研究的長讀數據中鑑定的轉錄亞型。從上到下的軌跡:IMMT基因座周圍 GENCODE 轉錄本的摺疊視圖、IMMT GENCODE 轉錄本的擴展視圖、突變體和 WT 的短讀覆蓋、短讀數據中推斷的突變體和 WT 之間的 delta-PSI,以及已知(綠色)和在長讀數據中識別的新(紅色)轉錄本。GENCODE 轉錄本類型用紅色(蛋白質編碼)、藍色(處理過的)和金色(哈瓦那合併)進行顏色編碼。編碼區域由 GENCODE 註釋軌跡中的填充顏色突出顯示。d RBM20中IMMT同種型表達的並排比較R634Q、P633L 突變體和 WT。兩種新的紅色轉錄本同工型IMMT-SL1和IMMT-SL2僅在 WT 樣品中表達。已知的 GENCODE 轉錄本為綠色,原始長讀為藍色。錯誤拼接的外顯子 6 用星號表示。IMMT-205和IMMT-208由於讀取計數低(所有樣本中為 1)而被省略。源數據作為源數據文件提供。
全尺寸圖片值得注意的是,並非所有具有差異表達轉錄本同種型的 107 個基因在總基因表達水平上都有顯着變化,如使用標準差異基因表達分析評估的那樣(方法部分和圖 4b))。在 107 個基因中,27 個表達單一同種型,其餘 80 個具有多種同種型的基因中,69 個基因顯示總基因表達差異。在這 69 個基因中,60 個基因在主要同種型表達上存在差異(> 基因總轉錄本的 50%),而 9 個基因僅差異表達次要同種型。具有多種同種型的 11 個基因的總基因表達沒有變化,但差異表達了一種或多種同種型。在評估轉錄本異構體的使用時,這 11 個基因中有 8 個顯示突變體和野生型之間異構體比率的顯着差異(Fisher 精確檢驗,調整後的P值 < 0.005,方法部分和補充圖 20),清楚地表明剪接水平的複雜調控,獨立於啓動子或同種型穩定性差異,因為總基因表達不受影響(補充表 5)。這種複雜的同種型調控表明,在基因水平而非同種型水平上測量差異 RNA 表達可能無法檢測到特定同種型的顯着表達變化,不需要事先註釋的方法是有利的。具有特定轉錄本同種型差異表達(而非總基因表達)的基因的兩個突出例子是TPM137和IMMT38,之前報告説兩者都是從大鼠和/或人類心臟轉錄組的短讀測序中錯誤拼接的。我們的長讀長數據清楚地表明,並非所有同種型在突變體中都受到同等影響(圖 4b)。對於IMMT,我們對短讀數據外顯子包含(PSI)分析的百分比確定僅外顯子6與野生型和突變體之間的差異剪接的,如在以前的研究中報道的37,38,41,42(圖 4C和補充文本)。然而,在全長轉錄亞型誤剪接的作用不是由以前的短讀分析捕獲的37,38。
具體來説,目前尚不清楚有多少和哪些轉錄本受到RBM20突變的影響。對於IMMT,基於參考 GENCODE 註釋,假設轉錄本IMMT-205是唯一受影響的同種型是合乎邏輯的,因為這是唯一沒有外顯子 6的IMMT註釋轉錄本。基於短讀數據的轉錄本量化差異表達分析確實將IMMT-205報告為IMMT(方法部分)的唯一重要影響。這是一種誤導,因為我們基於長讀長的方法可以清楚地識別兩種新的IMMT轉錄亞型,IMMT-SL1和IMMT-SL2,與它們相應的已知轉錄本IMMT-207和IMMT-204相比,缺少外顯子 6 。這兩種同工型在野生型中表達,在 R634Q 中不存在,僅在 P633L 中略有表達,與三種未差異表達的同工型形成對比(圖 4a、d)。在所有樣品中僅鑑定了IMMT-205 的一個讀數。基於域分析,我們發現外顯子 6 的缺失可能導致蛋白質具有新功能,因為該外顯子編碼的 32 個氨基酸的缺失破壞了蛋白質中一個現有的捲曲螺旋結構域和一個固有的無序區域(補充圖. 21 )。因此,新型IMMT的發現 isoforms 説明了無需事先註釋的全長轉錄組識別和定量的效用。
討論
總之,我們提出了人類細胞全長轉錄本亞型的全基因組分析及其在關鍵心臟基因RBM20中的剪接突變背景下的差異分析。雖然在剪接缺陷的RBM20突變37、38、40、41、42 中描述了不同的外顯子使用,但我們發現了選擇轉錄本上覆雜的異構體失調,這可能反映了啓動子活性、異構體穩定性和單個基因的可變剪接變化的複雜組合。在 RBM20 突變體中。我們的 FulQuant 工作流程允許直接識別和量化轉錄本異構體,無需事先註釋。以兩本小説為例IMMT轉錄本異構體是交替剪接的、無註釋的全長異構體測序,對於識別以前遺漏的許多新的、信息豐富的轉錄本異構體的存在至關重要。這些觀察結果,結合人類轉錄本異構體景觀的整體、複雜的複雜性(如圖 1 所示)和在基因組瀏覽器中),證明了更廣泛地採用長讀長測序進行轉錄組研究的迫切需要,以提供先前確定的可變剪接外顯子的背景。瞭解全長同種型對於理解表達基因的功能產物至關重要。我們直接量化和比較全長轉錄本同工型的工作流程,包括各類新型轉錄本,可以很容易地擴展到其他細胞類型和組織,以及已提議異常剪接在其中發揮作用的許多疾病。
方法人類 iPSC 中的基因組編輯
我們的出版物42詳細介紹了基因組編輯和 iPSC-CM 生成的方法。簡而言之,我們從斯坦福心血管研究所生物庫獲得了人類 iPSC。將退火(T4 連接緩衝液,NEB)和磷酸化(T4 PNK,NEB)引導 RNA(gRNA,使用http://crispr.mit.edu 上的工具設計)寡核苷酸引入 pSpCas9(BB)-2A 的 BbsI 位點-GFP 質粒並轉化到 STBL3大腸桿菌細胞中並通過 Sanger 測序驗證。將細胞以低密度鋪在基質膠包被的 6 孔板中,在 Essential 8 (E8) 培養基中接種一天後,將培養基更換為含有 Rock 抑制劑(Tocris Cat. No. 1254)的 E8。對於每個孔,CRISPR/Cas9 載體(1 μ,pSpCas9(BB)-2A-GFP)和 4 μg 單鏈 DNA 供體(補充信息)通過 Lipofectamine 3000 轉染引入細胞。轉染後 36-48 小時使用 FACSAria IIu(DB Biosciences)流式細胞儀與 100-μm 噴嘴分離 GFP + 細胞,用 Rock 抑制劑維持在 E8 中,用於前 3 天,初始密度為 2–3 × 10 3細胞/孔,隨後轉移到常規 E8,直到集落大小達到 ~0.5 mm。單獨的 iPSC 克隆被分離並重新接種到 E8 中 24 孔板的孔中,並含有 Rock 抑制劑。在目標基因組區域的 PCR 擴增後通過測序確認純合編輯(補充信息)。R634Q 電池可應要求提供。
iPSC-CM 生成通過調節 WNT 信號,iPSCs 分化為單層心肌細胞。iPSCs 以低密度接種在 Matrigel 塗層板上,使其在 4 天后(分化的第 1 天)達到 70-80% 匯合。用 3 ml RPMI 1640 (Life Technologies 11875-093) 和 1X B27® Minus 胰島素 (Life Technologies 0050129SA) 誘導分化,並補充 6 μM CHIR (TOCRIS 4953)。在第 6 天,將培養基更換為 3 ml 含有 1X B27® Minus Insulin 的 RPMI 1640,並補充有 5 μM IWR。從第 8 天起,將細胞保存在含有 1X B27® 無血清補充劑(Life Technologies 17504-044)的 RPMI 1640(Life Technologies 11875-093)中。從第 12-15 天開始,用含 1X B27 的 RPMI 1640 無葡萄糖(Life Technologies 11879-020)處理細胞,然後在含 1X B27 的 RPMI 1640 中恢復 2 天,並隨後以 3Mi 細胞/孔的密度重新種植。然後將細胞在含有 1X B27 的 RPMI 1640 中再保持 4 周,然後收集用於實驗。
逆轉錄使用 ZymoResearch RNA Clean and Concentrator-5 清洗和處理 RNA。總共 4.5 μL 混合物,含有 5 ng 純化的 iPSC-CM RNA,含 2.8% ERCC 和 3% Sequins A 版,1 μL 10 μM oligodT(/5SpC3/A*A*G*CAGTGGTATCAACGCAGAGGTACTTTTTTTTTTTTTTVTTTTTTTTTTTTTTTTTTTTTT1TTL dNTP、0.1 μL Takara 重組核糖核酸酶抑制劑 (40U/μL) 在 72°C 下孵育 3 分鐘,在 4°C 下孵育 10 分鐘,然後在 25°C 下孵育 1 分鐘。另一種 5.5 μL 混合物,包含 2 μL 5x SuperScript II 緩衝液、2 μL 5 M 甜菜鹼、0.5 μL 100 mM DTT、0.5 μL SuperScript II (200 U/μL)、0.25 μL Takara 重組核糖核酸酶抑制劑、0.1 μL 100 μM TSO、0.09 μL 無核酸酶水和 0.06 μL 1 M MgCl 2加入 10 μL 的最終反應體積。最後的 10 μL RT 反應在 42 °C 下孵育 90 分鐘,50 °C 下 2 分鐘和 42 °C 下 2 分鐘4進行 10 個循環。然後通過在 70°C 下孵育 15 分鐘並保持在 4°C 來停止反應。然後用 1 μL 1:10 稀釋的 NEB RecJf(目錄號 M0264S)在 37°C 下處理 30 分鐘,在 65°C 下處理 20 分鐘。
全長 cDNA 的 PCR 擴增將 25 μL 2x NEB Q5 HotStart 主混合物、0.25 μL 10 μM PCR 引物(ONT_Index1_ISPCR 和 ONT_Index2_ISPCR)5和 14.5 μL 無核酸酶水加入 RT 反應中。PCR 反應如下:98 ℃ 30 s,98 ℃ 10 s,67 ℃ 15 s,72 ℃ 6 min,72 ℃ 2 min,20 個循環。 4℃。PCR 反應使用 0.75 體積的 Ampure XP 珠進行純化,並用 25 μL 無核酸酶的水洗脱。使用高靈敏度 DNA 芯片在安捷倫生物分析儀上分析全長 cDNA。
牛津納米孔測序按照製造商的描述,總共將 1 μg cDNA 與 ONT 測序接頭連接,並在以下步驟中進行了修改: (1) 末端修復和 dA 拖尾;(2) ONT測序接頭的連接;(3) 添加測序系鏈。使用 MinION 設備上的 R9.4 和 R9.5 流通池進行測序。數據用 ONT 的 MinKNOW (1.4.2) 軟件記錄 48 小時。鹼基識別是使用 ONT 的 Albacore 軟件(版本 2.1.3)與選項“-disable_filtering -kit SQK-LSK108”和相應的流動池版本(補充表 1)進行的。
FulQuant 用於全長轉錄本異構體定量除非另有説明,否則分析是在 R (3.5.3) 中或使用自定義 Bash 腳本進行的。我們評估了對齊識別 (PID) 百分比對平均讀取質量(由 ONT 定義的 mean_qscore)的依賴性,並確定了平均讀取質量得分截止值 6,其中超過 80% 的讀取具有 PID > 80%(補充圖 2))。在去除低質量讀數後,我們在讀數的兩端(200 bp 窗口)修剪了測序接頭 ISPCR(AAGCAGTGGTATCAACGCAGAGTAC)和 polyA 序列(≥10 個連續 A/T,允許一個不匹配)。一端具有接頭序列而另一端具有polyA 序列的讀數被認為是全長的。我們使用 minimap2(版本:2.9-r751-dirty)和參數:’-K500m -secondary=no -a -x splice -splice-flank=yes’ 將讀數與人類基因組 (GRCh38) 對齊。從下游分析中刪除滿足以下標準的比對:(i) 由補充比對組成,(ii) 兩端的軟剪切大小大於 30 bp,(iii) 剪接點附近的插入大小(由雪茄 N 識別)> 5 bp 和 (iv) 未拼接對齊(單例)。
轉錄組註釋是對 WT 和 R634Q 的數據進行的。我們使用先前描述的聚類方法確定了內含子-外顯子和外顯子-內含子連接的標籤剪接位點45使用參數’HFWINSIZE = 5 DISTHRES = 8 PTHRESHOLD = 3 SUMHOLD = 5’。共享相同標籤剪接位點的讀數被摺疊成一致的轉錄本異構體。我們包括末端外顯子,但不考慮 5’ 和 3’ 端的轉錄變異,因為讀取兩端的錯誤率很高。事實上,對於末端外顯子,我們精確地註釋了它們的剪接位點,而不是它們的轉錄起始(TSS)和終止(TES)位點。我們使用一種轉錄本同種型中所有讀數的中位數 5’ 和 3’ 位置作為其 TSS 和 TES 位點。以下規則用於去除假陽性轉錄本:(1) 讀取計數 >3,(2) 全長讀取 >40%,(3) 轉錄終止位點 >10 bp 來自已知基因組 polyA 序列,(4) 鏈信息可用。此外,我們要求在每個基因座中的所有轉錄本:(1) 有超過 3% 的最豐富轉錄本的讀取計數,(2) 不是錯配的結果,以及 (3) 不是任何已識別轉錄本的 5’ 截斷產物,除非計數高於 25 % 較長的轉錄本。用於執行上述步驟的代碼附在 帶有詳細註釋的補充文件。對於最終的註釋,我們還刪除了只有 2 個外顯子或可能是已知轉錄本的截斷產物的轉錄本。
我們將我們的註釋與 GENCODE 人類綜合註釋 (V24) 進行了比較,以將轉錄本分類為已知的和新穎的。映射到已知 GENCODE 註釋的過濾轉錄本被拯救。我們進一步將新轉錄本註釋為新外顯子、新組合、外顯子跳躍、內含子保留、替代剪接位點和多個事件。可以在補充信息和代碼中找到每個新轉錄本類別的詳細定義 。
外顯子連接分析我們首先將所有轉錄本中存在的外顯子定義為組成型。對於基因內的所有外顯子對,我們計算了以下情況下的讀取數:(1) 兩個外顯子都存在,(2) 只存在外顯子 1,(3) 只存在外顯子 2,以及 (4) 兩個外顯子都存在不存在。使用這個計數矩陣,我們在使用BH 方法調整p值之後使用 Fisher 精確檢驗測試了它們的共關聯。遠端對被定義為至少相隔一個組成型外顯子的外顯子對。
預測無意義介導的衰變事件對於包含在 GENCODE 註釋中定義的已知起始密碼子的所有新轉錄本,我們計算了轉錄本上第一個終止密碼子和最後一個剪接點之間的距離。如果這個距離 >55 bp 46,我們將轉錄本定義為 NMD 產物。
短讀數據分析使用 STAR v2.5.1b 將讀數映射到 GRCh38 後,使用 featureCounts v1.6.0 確定基因表達水平。對於每個外顯子,我們分別將包含和排他的讀數計算為包括感興趣的外顯子的讀數和包括上游和下游外顯子但不包括感興趣的外顯子的讀數。計算了表示包含讀取與包含和排除讀取總和之間的比率的拼接 (PSI) 百分比。使用 Kallisto (0.46.2) 與 GENCODE 或我們的註釋作為索引的輸入註釋進行轉錄量化。
基因和轉錄本異構體水平差異表達分析通過計算共享該轉錄本相同剪接位點的讀數數量來實現轉錄本量化。通過總結該基因座中的轉錄物計數來實現基因定量。我們將 CRISPR 生成的 R643Q 和 P633L 突變樣本與未經 CRISPR 編輯處理的樣本 (WT_NC) 和未經 CRISPR 編輯處理的樣本 (WT) 進行了比較,其中使用 DESeq2 (1.26.0) 對基因和轉錄本進行標準差異表達分析異構體。由於測序深度有限,我們只考慮了所有樣本中總read數超過10的基因和轉錄本異構體來測試長讀長數據。使用 BH 調整的p定義重要候選者突變體和 WT 之間的比較值截止值為 0.001。對於短讀長數據,使用 DESeq2 的相同測試程序應用於使用 Kallisto 獲得的轉錄本和基因量化,無需任何過濾。GO富集分析使用PANTHER通過http://geneontology.org的網絡界面進行,表達的基因作為參考列表以控制背景。為了評估突變型和野生型之間轉錄本使用的差異,我們總結了生物學重複並對具有一種或多種差異表達同種型的 80 個候選基因進行了 Fisher 精確檢驗。使用 BH 程序調整P 值。
使用片段分析驗證新的剪接事件我們使用片段分析來驗證 33 個新的剪接事件(補充表 6和補充圖 10)。設計針對新剪接事件的寡核苷酸引物,使得具有和不具有剪接事件的擴增子將在凝膠上顯示可檢測的大小差異。所有引物序列都可以在補充表6 中找到 。我們進行了標準的 RT-PCR 並使用標準的 Bioanalyzer 分析分析了片段大小。只有包含具有預期大小的條帶的配置文件才被視為陽性驗證。對於 7 個不成功的案例,3 個案例沒有產生任何 PCR 產物,4 個案例包含與預測大小相差至少 20 bp 的片段。
報告摘要有關研究設計的更多信息,請參見與本文鏈接的 自然研究報告摘要。
數據可用性
本研究中生成的長讀長數據已存放在 ArrayExpress 中,登記號為E-MTAB-7334。本研究中使用的短讀取數據已存放在序列讀取存檔 (SRA) 中,登錄號為PRJNA57933642。我們的全基因組全長同種型數據集可在http://steinmetzlab.embl.de/iBrowser/ 上作為自定義基因組瀏覽器使用。 本文提供了源數據。
代碼可用性
GitHub https://github.com/czhu/FulQuant (doi: 10.5281/zenodo.4960275)上提供了執行轉錄過濾基本步驟的 R 代碼。
參考1.
王,ET 等。人體組織轉錄組中的替代異構體調節。自然 456 , 470–476 (2008)。
ADS CAS PubMed PubMed Central Article Google Scholar
2.Pan, Q., Shai, O., Lee, LJ, Frey, BJ & Blencowe, BJ 通過高通量測序深入調查人類轉錄組中可變剪接的複雜性。納特。基因 40 , 1413–1415 (2008)。