顏寧點評AlphaFold2 + 外行買家秀:蛋白結構預測神器初體驗_風聞
返朴-返朴官方账号-关注返朴(ID:fanpu2019),阅读更多!2021-07-27 10:13

時隔半年,7月16日,DeepMind在《自然》雜誌上發文,公開了進一步優化的AlphaFold2源代碼並詳細描述了其設計框架和訓練方法。幾天後,DeepMind又發佈了由AlphaFold預測的蛋白結構數據庫,免費提供給全球科研人員開發使用。對此,《返樸》總編、結構生物學家顏寧提出了自己的見解。
最近這次AlphaFold帶來的震撼其實不如第一次大,因為有了上一次的突破,現在這個結果基本是水到渠成。對比了一下鈉離子通道的結構——預測的部分與電鏡已經解析的部分吻合得還行,但沒有任何homology model(同源模型)的部分,比如電鏡結構裏看不到的部分,在預測的結構裏依然是一團無序的圈圈。
AlphaFold毫無疑問是對整個生物學的一場變革,而不侷限在結構生物學領域。但它並不是結構生物學的終點,而是可以把結構作為起點去做更多的東西。生命在於運動——分子層面的運動是冷凍電鏡技術革命之後結構生物學新的重點。以我的科研為例,我們的目標並不是要獲得研究對象的摺疊信息,而是要解析其處於不同工作狀態的精準構象信息,以此來理解它的工作機理和致病機理。
短短几天,學界針對AlphaFold已經形成兩派:做超級複合物的歎為觀止,做膜蛋白的,比如我,感覺幫助不大,並且誤差較大。這是為什麼?
有人認為,是因為膜蛋白有脂水兩相;有人認為,是因為膜蛋白本身解出來的可靠數據少,不夠供AI訓練;還有評論説,是因為膜蛋白靜態簡單,動態複雜;超級複合物則相反,而AlphaFold強在靜態蛋白結構預測。
膜蛋白的摺疊有着相對複雜的脂分子環境,一旦缺少可靠的模型,AlphaFold所預測的蛋白質摺疊可能大體正確,但在具體構象上與實驗數據有較大的差距。而決定膜蛋白複雜功能的,不是它相對簡單的摺疊模式,而是精細的構象變化。
AI最大的挑戰,將會是在未來。關於分子的動態研究、動態預測、動態模擬等等,其實還有很多問題都值得解決。在上次AlphaFold橫空出世的時候我就説過,希望AI下一步可以解決分子動力學模擬對於很多生物過程無能為力的問題。那就繼續期待AlphaDynamics吧?
——顏寧
撰文 | 繼省
最近一週以來,生物醫藥圈子被AlphaFold v2.0刷屏,每天打開微信朋友圈,都能看到至少一半的新消息與它有關。這款由谷歌旗下人工智能公司 DeepMind團隊開發的蛋白三維結構預測程序,在2020年的蛋白結構盲測比賽CASP中一騎絕塵獨領風騷;而這次研發團隊把全部代碼任性地開放,並把Uniprot*上大部分代表性蛋白序列都給預測了一遍。
*UniProt是一個免費使用的蛋白質序列與功能信息數據庫。
與此同時,蛋白設計領域的大神David Baker課題組參考AlphaFold的思路,利用他們長期在蛋白質設計領域的優勢推出了一款同類軟件RosettaFold。後者雖然不如AlphaFold覆蓋全面,但在一些代表性的蛋白結構預測上已經可以與之媲美,並且對計算資源的佔用更少。
我偶爾也會八卦:是不是RosettaFold的良性競爭壓力促成了AlphaFold的全面開源?總之,一時間結構生物學、人工智能、合成生物學還有生物醫藥投資等多個領域的研究者開始了熱烈的討論。
但熱鬧是他們的,我只關心我課題裏的蛋白能不能利用這兩個程序的某一個來凹個造型,哦不對,預測個結構。
對於絕大多數實驗生物學從業者來説,我們在實驗室中研究某個蛋白功能之後,常想進一步瞭解一下它們的空間結構,這樣就能夠更好地理解和詮釋我們在實驗中看到的一些現象,也可以針對空間結構去開發設計一些藥物,來阻斷蛋白的功能。——當然,這部分設計需要另外的專家。
AlphaFold把預測結果以數據庫的形式公開在了網絡上。對於只想薅羊毛的用户,不啻天外福音。於是我興致勃勃地打開了它的預測結果查詢網站(alphafold.ebi.ac.uk)。
好,先來一個試試。以前讀博士時,經常研究一種叫beta-catenin的蛋白,這是一個在脊椎動物個體發育和癌症等多個生物學過程裏都非常重要的蛋白。輸入蛋白名稱後,得到25條結果,對應人、大鼠、小鼠、斑馬魚等多個模式物種。這25條當然不是現在地球上已知叫beta-catenin的全部蛋白,但能有模式物種的結果,就很有代表性了。



但是預測出來這個犰狳重複並不出奇——PDB數據庫*現已發表數個人類beta-catenin結構,説明這段序列比較穩定,換言之就是供AlphaFold學習訓練的知識比較充沛,它預測也比較容易。反倒是這段重複區域的前後兩端(N端和C端)還各有近百個氨基酸,至今沒有穩定的結構問世(當然,可能蛋白本身在這個區域就屬於比較混沌無序的),而AlphaFold的模型裏,在N端(氨基端)和C端(羧基端)也同樣分數很低,並沒有顯著改善。
*PDB,全稱Protein Data Bank,是目前最主要的收集蛋白質三維結構的數據庫。
再試驗一個我現在導師課題組裏研究多年的膜蛋白TGFBR2,這是一個受體酪氨酸激酶,既是膜受體又是激酶,同樣沒有全長結構問世。同樣的流程,找到人TGFBR2,打開結構後是下面的情形:這個蛋白明顯出現了三個分數比較高的區域,包括靠近N端的配體結合區,中間的跨膜區,和C端的激酶區,從序列上看,和目前人們對這一蛋白的功能認識很吻合。而畫面中橙黃色低分區域,也同樣是PDB已有結構裏缺失信息的部分。
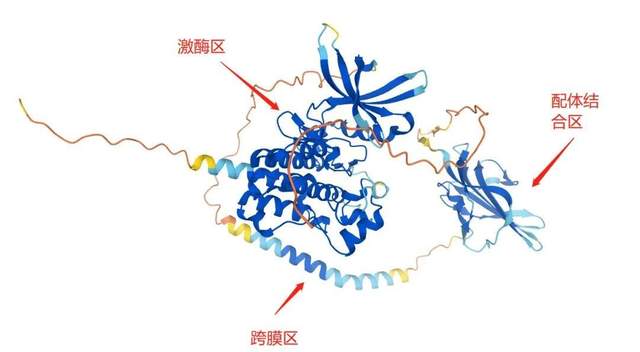

當然了,説一模一樣太誇張,還是有細微差別的。最重要的一點,右側實驗得到的結構裏有一個化學小分子結合到蛋白上,這是一種生理調控下的蛋白狀態;蛋白在有無化合物結合、不同化合物結合的狀態下都會呈現不同的細微變化,而正是這些變化才體現了蛋白質的多樣而神奇的功能。在這一點上,AlphaFold給出了一個單一的最優解,是無法覆蓋蛋白質的千姿百態的。
但即便如此,AlphaFold也是做到了此前人們無法想象和做到的事情。
做了兩個測試之後,自然就想到,如果隨意給一段序列,能否輕鬆地得到一個結構模型呢?畢竟這才是AlphaFold最讓人興奮的地方。AlphaFold的正式運算需要的資源非常龐大,個人或者小團隊如果家裏沒礦的話,想自己搭平台運算基本沒戲;好在谷歌提供了一個免費的雲端平台Colab(https://colab.research.google.com/github/deepmind/alphafold/blob/main/notebooks/AlphaFold.ipynb),可以運行精簡版的AlphaFold。平台入口就在AlphaFold數據庫的底端FAQ裏。
這個雲端平台解放了硬件,只要你準備好了要預測的氨基酸序列就可以了。當然運算方法比完整的AlphaFold要“簡陋”一些。但聊勝於無,要啥自行車呢?
結構預測前的準備工作有三步:第一步連接平台,第二步安裝第三方軟件,第三步下載AlphaFold。連接平台需要登錄(似乎只能用谷歌賬號,不太確定國內的同行們能否使用),安裝軟件和下載AlphaFold只需點一下鼠標,各需2-3分鐘。當真是哪裏不會點哪裏,So easy!



在AlphaFold搜庫的半小時裏,我還探索了一下RosettaFold。華盛頓大學David Baker實驗室課題組搭建並公開的這個服務器每週都能吸引超過5000條計算任務,我用Colab上的示範序列來測試RosettaFold的預測效果,提交了任務之後發現前面還有3000個任務在排隊,且等着吧。從運算時效上來看,還是AlphaFold Colab更快。有意嚐鮮且手裏有實驗數據的朋友,不妨兩個平台都試試。
前面我在AlphaFold數據庫裏檢索的beta-catenin和TGFBR2兩個蛋白,其實屬於已知結構的蛋白了,Uniprot上有大量尚無PDB結構的蛋白條目,在AlphaFold上都有預測結果。比如AlphaFold數據庫首頁搜索框下方的示例Q8W3K0,這是一種存在於模式植物擬南芥裏的蛋白,可能具有抗病作用。AlphaFold預測出來的結果驚豔到我了,蛋白好漂亮!難怪無數俊男美女投身到蛋白結構解析的大軍中,這真的是門藝術。
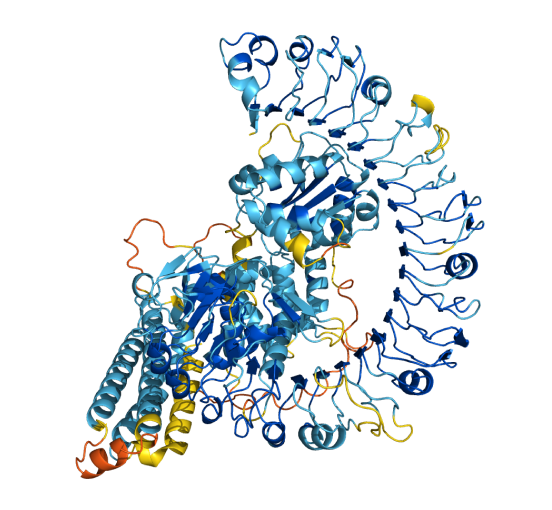
以上就是我簡單的體驗經歷。從我不成熟也不算專業的角度來看,AlphaFold對結構生物學家的衝擊可能不大:一來,蛋白的無序區域(指不能形成穩定三維結構的區域)涉及氨基酸分子的自由運動,箇中規律仍然需要結構生物學家去揭秘;二來,很多重要的生理病理性蛋白(如離子通道蛋白),在不同的活性狀態下都會有很精妙的構象變化(conformational change),在細胞內外也會因為和各種各樣的其他蛋白結合而呈現出千變萬化的空間構象。以AlphaFold目前僅用蛋白本身序列作為輸入信息的算法邏輯,至少還不能熟練應對上面提到的幾種情形。
但AlphaFold的問世,本身就是一件打破常規超乎想象的創舉,我們有理由期待這個工具會不斷進化,持續給我們驚喜。如顏寧老師所説,也許在不久的將來,隨着人們對蛋白質分子動力學知識的加深,引入分子動力學模擬的AlphaDynamics會橫空出世,來預測出同一個蛋白的多個穩定構象。而對廣義的實驗生物學家來説,AlphaFold無疑提供了一個全新又容易上手的虛擬驗證平台——設計一款融合蛋白,一個全新的小肽,或是給自己中意的蛋白製造幾個氨基酸突變,拿AlphaFold的預測結果和實驗結果相互佐證,會讓本就刺激的工作更加有趣,也可以為AlphaFold未來的進化提供了更多真實數據。
未來可期!願大家摺疊快樂!