司長退休後,拍了一組“罕見”照片,呼籲更多人關注_風聞
医学界-医学界官方账号-为你提供可靠、有价值的内容是我们的存在方式。2022-02-28 21:44
“他們有與我們不同的生理狀況,但與我們有相同的生命,請關注他們,完善我們的社會制度。”
一場主題為**“關注”的攝影展,正在上海舉行,鏡頭對準的是罕見病患者**。
拍下這些照片的攝影師,是國家醫療保障局醫藥服務管理司原司長熊先軍。退休之前,攝影是他的愛好,退休之後,攝影成了他的專業。

“總想用攝影寫一部關於‘生’的書——生命的生,但不知如何下筆。”熊先軍講述了為什麼選擇拍攝罕見病羣體,2021年,他參加了中國罕見病聯盟舉辦的罕見病大會,讓他找到了關於‘生’的切入點。
微小的基因差異,造成了罕見病患者生而不同。這個不同是基因差異造成的他們生命的差異,還是我們整個社會制度還不能包容罕見病患者,而造成了他們生命的差異?熊先軍説:“我看答案是後者。”
熊先軍希望通過他的攝影,呈現給公眾一個具體的罕見病患者的形象,以引起更多人關注罕見病,關注罕見病羣體。“他們有與我們不同的生理狀況,但與我們有相同的生命,請關注他們,完善我們的社會制度。”

致命的“舞蹈”,特別的心願
亨廷頓舞蹈症是一種罕見的常染色體顯性遺傳病。一般在中年發病,出現運動、認知和精神方面的症狀。臨牀特點有不由自主的舞蹈樣動作,精神行為障礙和認知功能損害。目前治療該病的藥品為氘丁苯那嗪片,已納入醫保。
韓愛華 58歲,已確診22年。從1997年開始出現情緒不穩、寡言少語,有抑鬱傾向,並逐漸出現身體不協調、走路常跌倒、無法説話等症狀。已卧牀11年,失去意識,不認識人。20年多來,丈夫一直無微不至地照料着妻子。他們有一個心願,希望在愛華去世後將遺體捐獻給科研機構,為治療亨廷頓舞蹈症做貢獻。目前家庭月收入4500元,無低保。與兒子同住。患者已不服用治療亨廷頓的藥,只吃治感冒等常見病藥,報銷約90%。家庭年醫療費大約幾千元。

“害怕”蛋白質的寶寶
甲基丙二酸血癥 又稱甲基丙二酸尿症,是我國最常見的常染色體隱性遺傳的有機酸代謝病。由甲基丙二酰輔酶A變位酶或其輔酶鈷胺素代謝缺陷所導致。據估算,我國大陸地區出生患病率約1/28,000,北方有些地區可高於1/10,000。通常發病年齡越早,急性代謝紊亂和腦病表現越嚴重。新生兒期發病者多在生後數小時至1周內出現急性腦病樣症狀,病死率高。兒童期發病者多在1歲以內,如不及時治療,可導致智力發育和運動發育遲緩、落後和倒退,可伴發血液系統、肝臟、腎臟、皮膚和周圍神經受累。成人患者首發症狀可為周圍神經病變和精神心理異常等。目前主要治療方案為長期食用特食和輔助代謝類藥物。
於竣亦 3歲半,出生十多天確診,與父母和姥姥同住。家庭沒有穩定收入,患者爸爸現在只是打零工,媽媽在家照顧孩子。患兒目前食用特奶、特食。服用藥物左卡尼汀、精氨酸和穀氨酸,因是超適應症用藥,醫保不報銷。患者發育遲緩,平時定期接受康復訓練。患者月花費5,000元左右。

愛心給我力量
杜氏肌營養不良 是一種X染色體隱性遺傳疾病,主要發生於男孩。據統計,全球新生男嬰發病率為1/3500。患者在學齡前就因骨骼肌不斷退化導致肌肉無力或萎縮,行走困難。到12歲之前會徹底喪失行走能力,20多歲左右會因心肌、肺功能衰竭而危及生命。
胡星鑠 13歲,5歲半確診,和父母一起生活。患者口服激素藥物和接受康復鍛鍊。母親在家陪護患者,爸爸工作,一個月收入1.5萬元,房租一個月1200元,醫藥費一個月2000元左右,康復一次200多元,每年複查後去康復科調整康復方案,再回家自行鍛鍊。由於患者是山東醫保,醫藥費用在北京不能報銷。

“硬皮病”遮不住美麗笑臉
系統性硬化症也稱為硬皮病,是一種自身免疫性結締組織疾病,確切致病原因尚不明確,現有研究認為和血管病變、自身免疫異常、皮膚和內臟器官廣泛纖維化有關。此病多見於女性,易導致患者容貌發生改變和肢體殘疾,累及內臟時會危及患者生命。目前尚無特效藥物。
鄭嬡 31歲,1998年出現症狀,皮膚緊繃、身上有白斑出現,後來被確診。高中時,臉上出現紅點、嘴唇變薄。上大學後病情略有嚴重。大學畢業後,因病求職困難,於2016年創辦了硬皮病患者組織,成為全職為病友服務的公益人。現主要服用激素、中藥、鈣片等,每月醫藥費2000元,自己負擔。硬皮病患者多為女性,且影響容貌,會引發自卑、抑鬱等心態。鄭嬡告訴病友:每一位硬皮病患者的笑容都很美,我們的人生,可以很燦爛。

相伴“肯尼迪”的“遊俠”
脊髓延髓肌萎縮症又稱肯尼迪病,是一種遲發的X-連鎖隱性遺傳性神經系統變性疾病,主要累及下運動神經元、感覺系統和內分泌系統,逐漸剝奪患者的行動能力、吞嚥能力和呼吸能力。患病率為1-2/10萬,僅男性發病,多見於30~50歲。目前尚無有效治療方法。
劉期達 2007年出現下肢乏力症狀,曾被誤診,後在北京宣武醫院確診。在發病14年之後,他已經在蹲下後無法自行站起,上樓梯非常艱難,連續步行的能力大幅度下降,每走100多米就必須停下休息;由於面部肌肉萎縮,無法自然地展示笑容。目前每月醫藥費2000元左右,無法報銷,收入上可以負擔。十年來,他趁着還有行動能力,自駕旅行周遊全國,於2014年發起成立“肯尼迪罕見病關愛中心”,通過自媒體平台為病友送上積極對抗疾病的信心。

無汗少汗、四肢劇痛的人生
法佈雷病是由於患者GLA基因發生突變,導致體內α-半乳糖苷酶A活性降低或完全缺乏,造成人體代謝底物酰基鞘氨醇三己糖及其衍生物脱乙酰基鞘氨醇三己糖在患者血管、神經系統和各臟器貯積,對腎、心臟、腦、神經等各器官產生嚴重損害。病情呈進行性加重發展態勢,如得不到有效治療將危及生命。該病以X性連鎖方式遺傳,男性症狀通常比女性嚴重。目前特效治療藥物為注射用阿加糖酶ß、阿加糖酶α,年治療費用在100萬元。
耿京博 34歲,2014年轉診多家醫院,按慢性腎炎治療,2015年通過腎穿刺確診。現在四肢疼痛的時候吃止疼藥,腎臟方面吃一些降低尿蛋白的輔助藥物。因特效藥太貴,沒有使用,無法從根源上解決疾病問題。夫妻與孩子及爺爺奶奶同住,夫妻每月收入1.4萬元,爺爺奶奶還有部分收入,每月醫藥費報銷後自費500-600元。他希望能將特效藥納入醫保,每年個人負擔不超過5萬元。
我們的心永不“漸凍”
肌萎縮側索硬化症屬於運動神經元疾病,是上運動神經元和下運動神經元損傷之後,導致包括球部(是指延髓支配的肌肉)、四肢、軀幹、胸部腹部的肌肉逐漸無力和萎縮,患者俗稱“漸凍人”,國內治療藥物為依達拉奉氯化鈉注射液,已納入醫保。
金冬浩 44歲,2019年12月確診,在確診之前經歷了多家醫院就診的經歷。現在患者沒有工作在家,和父母同住,靠儲蓄生活,父母照顧他。患者有內服和外用藥物,每月花費3800元,醫保報銷2/3,自費1400元左右。女兒和愛人平時在北京西城區居住,因為女兒在那上學。

“月亮孩子”也能很陽光
白化病是由於人體黑色素在合成和加工過程中受到減弱或缺失而出現的一種遺傳性病症。患者眼部色素缺失,毛髮白或黃栗色,皮膚白皙,且終身如此。該病以眼損害最為明顯,多數病人視力低下、嚴重怕光等,且無法矯正;皮膚極容易被日光中的紫外光曬傷,較易產生皮膚癌。據估算發病率約為1/15,000,我國約有10餘萬人。沒有特效治療藥物。
謝航程 28歲,一出生就確診,姐姐也是。他自幼就知道,要避免暴曬來保護皮膚,視力較弱,常俯身看東西,對生活其他方面影響不大,不需要針對性的治療。他早年學習過嗩吶,2017年來北京,在打工子弟學校當音樂教師,同時加入由罕見病病友組成的8772樂隊,擔任吉他手,也做兼職的吉他老師。音樂讓航程擁有了面對生活的自信與陽光,在舞台上演奏着自己的青春激情。

當生活被“大手大腳”改變
肢端肥大症是腺垂體分泌過多生長激素所致的面容變化和內臟器官異常肥大,並伴有相應生理功能異常的內分泌代謝疾病。生長激素和生長因子過多主要引起骨骼、軟組織和內臟過度增生,在青春少年表現為巨人症(gigantism),在成年人表現為肢端肥大症,嚴重者可出現心力衰竭和睡眠呼吸暫停等合併症。
劉剛 38歲,畢業於航空院校,曾為飛行員,因病中斷,成為一名地面空管。2007-2008年在北京協和醫院確診後,先後經歷兩次手術,現在激素值比正常高一些。一年隨診一次,沒有服用特別的藥物,換季時喝中藥調節血壓,醫藥費可報銷,自費較少。他有着幸福美滿的家庭,愉快又辛勤忙碌的工作,以及穩定的收入。

與腫瘤平和相處
神經纖維瘤病一種良性的周圍神經疾病,屬於常染色體顯性遺傳病,常累及如神經系統、眼和皮膚等,是常見的神經皮膚綜合徵。主要的致病機制為腫瘤生長對周圍組織的破壞,如消化道出血等;腫瘤增長對周圍神經的壓迫導致麻木、肌無力等;腫瘤於顱內因佔位效導致顱內壓增高產生頭痛、嘔吐等症狀;或刺激腦組織異常放電形成癲癇等。患者需面臨經常性手術切除各部位腫瘤。目前沒有特效治療藥物。
鄒存偉 50歲,小時候症狀不明顯,直到腰椎上長瘤並疼痛不已時,才開始就醫,1996年確診,接受了手術。在此後20多年裏,又做了3次手術。最近一次手術,他切除了引起腸梗阻的腫瘤,花費了1萬多元。手臂上的一顆腫瘤,讓他手臂酸脹無力,除此之外,對平常工作和生活影響不大。無治療,無用藥。目前單身,獨自生活,個人年收入税後9萬左右,居住在公租房。

期待展翅高飛的“蝴蝶寶貝”
大皰性表皮鬆解症一種罕見的遺傳性皮膚病,其臨牀特徵是皮膚或黏膜受到輕微摩擦後出現水皰或血皰,進而皮膚創傷、潰爛。重型的患者因反覆瘢痕形成可能出現肢體殘毀、活動受限,手指腳趾粘連及食道狹窄等情況。因為皮膚像蝴蝶翅膀一樣脆弱,且多數從出生起即發病,病患被稱為“蝴蝶寶貝”。口服和外用抗感染藥物可以預防創面的繼發感染,傷口護理採用水膠體或泡沫敷料加快癒合。目前無特效治療藥物。
高歆一茸 10歲,出生就被診斷,2014年做基因檢測進一步確診。現在有貧血症狀,體型較瘦,營養不良。一茸因病一直無法上學,尤其夏天傷口較多,怕曬,不能出門,只能在家上網課;冬天隨着氣温降低、乾燥,身體情況稍有緩解,可以參加線下興趣班。目前媽媽全職在家照顧一茸,主要靠爸爸工作,年收入10萬餘元,老人可幫補一些。醫療支出每月2000元左右,主要是團購國外進口的敷料,醫保不報銷。
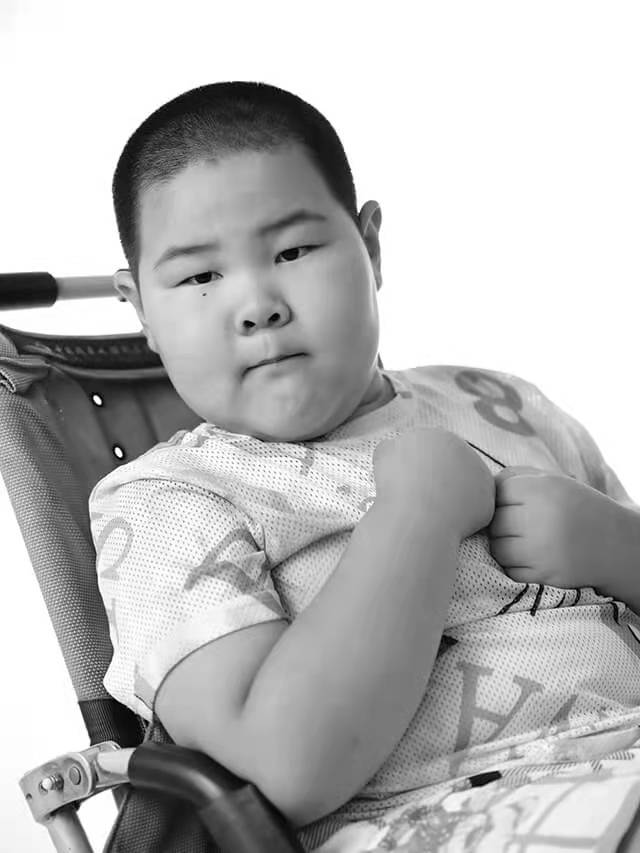
輪椅上的微笑
脊髓性肌萎縮症 脊髓性肌萎縮症是一類由脊髓前角運動神經元變性導致肌無力、肌萎縮的疾病。臨牀表現差異較大,根據患者起病年齡和臨牀病程,將SMA分為4型,1型最為嚴重。目前已有特效藥諾西那生鈉注射液、利司撲蘭口服溶液用散在中國獲批上市。年治療費用在200萬元左右。
周建濤 9歲,出生一週的時候確診SMA2型,目前坐輪椅,居家康復。父母平時在北京市內上班。因特效藥治療費用昂貴,患者無任何治療。患者和爺爺奶奶一起生活,爺爺奶奶在家種地打點零工,並照顧患者。

“瓷娃娃”一樣堅強
成骨不全症 是以骨量低下、骨骼脆性增加和反覆骨折為主要特徵的單基因遺傳性骨病,可有藍鞏膜、牙本質發育不全、聽力異常、關節韌帶鬆弛和心臟瓣膜病變等骨骼外表現。多數呈常染色體顯性遺傳,少數呈隱性遺傳,罕有X染色體伴性遺傳,是骨骼中型膠原蛋白數量減少或結構異常相關的疾病。
王琳 34歲,出生18天在沒有摔倒的情況下骨折,8個月時確診。此後骨折十餘次,成長過程中沒有接受特別的治療或康復方案。因為疾病影響,從小無法和其他小朋友一樣上學,曾長期沒有出過家門。她曾只能卧牀,經鍛鍊可以坐輪椅出行、拄拐站立。平時服用鈣爾奇D補鈣,沒有進行其他治療。她作為大連殘疾青年協會主席,組織了多項公益活動。她丈夫也是成骨不全症患者,目前二人同在北京從事罕見病公益支持工作,為廣大病友奔走“吶罕”。

“黏寶寶”的多彩人生
黏多糖貯積症 一組複雜的進行性多系統受累的溶酶體病,由於降解黏多糖的酶缺乏,不能完全降解的黏多糖在細胞中貯積,可造成面容異常、神經系統受累、骨骼畸形、肝脾增大、心臟病變、角膜渾濁等多種症狀。常見一共有7個分型,目前部分分型有特效藥物治療,但價格十分高昂。目前特效治療藥物有:目前特效治療藥物有:依洛硫酸酯酶α、拉羅尼酶、艾度硫酸酯酶β,年均治療費用在100萬元左右。
顧若凡 21歲,2004年確診黏多糖貯積症Ⅳ型,目前在持續用藥。自幼因病去了很多地方求醫,也讓若凡有了遠超同齡孩子的閲歷。2019年,若凡成功申請到美國大學學習平面設計,也因此終於可以用上夢寐以求的特效藥,藥費大概需要45萬美元,美國醫療保險全部可以報銷。異國求學中,喜歡畫畫的她用才藝得到了同學們的讚歎,也讓她有勇氣在藝術的道路上不斷耕耘,曾登上《中國夢想秀》的舞台,用沙畫演繹追夢人生。目前因疫情暫時中斷學業回到國內,和媽媽一起生活。家庭收入一年接近20萬元,在國內一年足量用藥的治療費用要在300萬元左右,若凡只能依靠公益援助和自費盡力保障當前的持續用藥。

惟盼晝不眠、夜無夢
發作性睡病一種原因尚不明確的慢性睡眠障礙,臨牀上以不可抗拒的短期睡眠發作為特點,在我國多於兒童或青少年期起病。表現為白天過度嗜睡、猝倒、睡眠癱瘓、入睡前幻覺和夜間睡眠紊亂。
暴敏冬 37歲,2008年發病,2014年確診為發作性睡病。日常嗜睡和猝倒都比較嚴重,因病患上了抑鬱症。如今的她能夠與疾病和解,接受自己的身體現狀,也已適應現在的生活。偶爾會焦慮,但整體樂觀。做過記者、保險顧問,現全職負責發作性睡病的病友組織,為廣大病友提供醫療信息、心理支持並呼籲社會各界關注發作性睡病羣體,消除對這一羣體的歧視。現前往博鰲樂城試用藥物,一個月2萬元,來回花銷1萬元左右,但效果沒有達到預期,服用了2個多月,尚未報銷。現階段國內沒有針對此疾病的藥,患者平時就抑鬱症和多動症的診斷開藥,同時用其他藥物控制猝倒。

美好從突破自我開始
神經母細胞瘤是兒童最常見的顱外腫瘤,有將近一半的神經母細胞瘤發生在2歲以內的嬰幼兒。屬於神經內分泌性腫瘤,可以起源於交感神經系統的任意神經脊部位。其最常見的發生部位是腎上腺,但也可以發生在頸部、胸部、腹部以及盆腔的神經組織。目前已知有少數幾種人類腫瘤,可自發性地從未分化的惡性腫瘤退變為完全良性腫瘤,神經母細胞瘤就屬於其中之一。目前沒有特效藥物。
潘美好 27歲,一歲時因突發高燒被確診。腫瘤壓迫了椎管運動神經,歷經多次化療,渡過生命危險,但因脊髓損傷無法行走,從小到大與輪椅相伴。10歲時因身體原因輟學在家,在2016年參加了北京的“自立生活”公益項目,逐漸可以在北京獨立生活,自在地參與公益活動、找到了能保障收入的工作,還曾嘗試過跳傘、潛水、攀巖等。目前,她和病痛挑戰基金會一起,參與神經母細胞瘤患者社羣的公益賦能工作。她喜歡稱呼自己“輪椅上的好姑娘”,期待用自己的故事,給更多殘障夥伴以勇氣和信心。

告別“大肚子”的心願
戈謝病是一種因溶酶體中葡萄糖腦苷脂酶功能缺陷導致的罕見常染色體隱性遺傳代謝病。由於機體葡萄糖腦苷脂酶活性缺乏或降低,造成其底物葡萄糖腦苷脂在肝、脾、腎、骨骼、肺、甚至腦的巨噬細胞中貯積,形成典型的貯積細胞即"戈謝細胞",導致受累組織器官出現病變,臨牀表現為多臟器受累並呈進行性加重。特效藥;伊米苷酶、維拉苷酶a,根據體重不同年費用在70萬元到200萬元。
高子博 13歲,2014年確診戈謝病三型。因病脾臟不斷腫大,肚子一天天鼓起來,又用不起藥,2015年為了保命,不得不做了脾切除手術。2016年因切脾,患者出現股骨頭壞死的現象,走路有點跛,肝也逐漸增大。2021年有幸參加了戈謝病藥物臨牀試驗,臨牀可以免費用藥1年,每半個月從河北來北京用藥一次,來回費用大概幾百元,臨牀試驗可報銷200元。另外患者還服用一些其他輔助藥物。子博的弟弟在兩年前也確診了戈謝病。媽媽在家照顧孩子們,爸爸在印刷廠工作,月收入僅4000元左右。家人現在擔心子博用藥1年後如果斷藥,身體惡化會更嚴重,盼望兩個孩子的肚子不要一天天鼓起來……

袖珍人的廣闊天空
垂體柄阻斷綜合徵是一種由於垂體柄缺如或變細並垂體後葉異位,下丘腦分泌的激素不能通過垂體柄輸送到垂體後葉,繼而不能作用於垂體前葉而導致的垂體功能減退的症狀。估計發病率為1/200,000。其導致的激素分泌異常包括:從孤立性生長激素缺乏到合併其它多種垂體前葉激素缺乏,部分患者還同時存在垂體後葉激素缺乏,呈現出尿崩症表現。童年期表現為身材矮小、低血壓等;青春期,第二性徵不發育。目前,模擬生理性內分泌激素分泌的替代治療,可重建垂體功能,讓患者生長、發育,甚至“自然地”生育。主要治療藥物為重組人生長激素,在醫保目錄中限制為兒童原發性生長激素缺乏症。
袁納納 38歲,身高定格在142釐米。長期使用生長激素、甲狀腺激素、腎上腺激素、性激素替代治療,年治療費用大約為6-8萬,醫保報銷2000元,剩餘部分完全自費。她在北京從事NGO的工作,收入不穩定,主要靠家人支撐醫療費用。她是北京市東城區袖珍人之家發起人,近10年來,通過殘障女性領導力培訓、推動政策研討、一系列公眾倡導等工作,努力改變社會對生長發育障礙羣體的偏見,鼓勵病友自由勇敢地成長。

陽光樂觀的“小飛俠”
面肩肱型肌營養不良是一種以進行性加重的肌肉無力和萎縮為特徵的遺傳性神經肌肉疾病,為常染色體顯性遺傳,通常在青春期或成年期發病。患病率為1/2萬。最常受影響的肌肉組織為面部、肩胛骨與上臂,並且隨着疾病加重影響身體軀幹和下肢的肌肉羣。嚴重患者會出現吞嚥、眼底部、高頻聽力、認知、説話障礙、呼吸功能方面等問題。約有20%的患者最終需要用輪椅。目前暫無任何治療方法。
肖可心 22歲,2017年因手臂不能上舉手被確診。初中時期腰腹無力不能完成仰卧起坐,高中時期左臂不能正常舉起,背部呈現“翼”狀;右手手指不同程度萎縮;長期以來,不能正常吹響哨子或氣球、不能正常通過吸管吸取液體;在睡眠期間,不能完全閉合雙眼;會出現肌肉中的“不適”或“灼痛”的症狀。由於堅持鍛鍊,她讓症狀維持在以上表現,順利完成了學業,在北京開啓了工作後的嶄新人生。

與失明、癱瘓的威脅時刻相伴
多發性硬化是以中樞神經系統白質炎性脱髓鞘病變為主要特點的自身免疫病。本病最常累及的部位為腦室周圍白質、視神經、脊髓、腦幹和小腦,主要臨牀特點為中樞神經系統白質散在分佈的多病灶與病程中呈現的緩解復發,症狀和體徵的空間多發性和病程的時間多發性。如不及時診治,可造成患者殘疾(失明、癱瘓等)。可通過藥物治療減少疾病復發並控制病情發展,不同情況的病友適用於不同的治療方案。目前主要治療方案為疾病修正治療(DMT),大多數藥物已經納入國家醫保,但仍存在落地困難。
秦巖鵬 33歲,2018年3月第一次發病,右眼視野部分丟失,去眼科就診,2018年4月確診多發性硬化。確診初期沒有用藥,2018年10月開始服用一種特效治療藥物,當時未進醫保。目前仍在用藥,醫保報銷後每月花費2000元左右,另外每週進行2-3次中醫針灸,醫保報銷後每月花費400元左右。患者收入一個月約4000元。

“自毀容貌”症兒童的微笑
Lesch-Nyhan綜合徵 即自毀容貌綜合徵,是X連鎖隱性遺傳的先天性嘌呤代謝缺陷病,源於次黃嘌呤一鳥嘌呤磷酸核糖轉移酶缺失,使得次黃嘌呤和鳥嘌呤不能轉換為IMP和GMP,而是降解為尿酸,出現高尿酸鹽血癥。見於男性,患者表現為尿酸增高及神經異常,如腦發育不全、智力低下、攻擊和破壞性行為。有咬傷自己的嘴唇、手和足趾等強迫性自身毀傷行為。目前尚無特效治療辦法。
果果 2014年出生,特別愛笑,百天照都是笑眯眯的,但已經有了仰頭和身體弓形等肌張力高表現。5個月時發現不會翻身,曾被誤診為腦癱。14個月時確診。他會不受控制地自殘,1週歲開始反覆咬下嘴唇,生生把下嘴唇咬掉了,後來又咬手,為了保住手指等,家人只能把他的牙拔掉。現在有明顯的肌張力障礙和智力障礙。他喜歡看動畫片,不時露出可愛的笑臉。
注:照片使用已獲得本人及出品方授權。本文疾病和患者介紹文案由病痛挑戰基金會提供。