有效性可疑,安全性堪憂,抗新冠口服藥阿茲夫定是怎麼上市的?_風聞
返朴-返朴官方账号-关注返朴(ID:fanpu2019),阅读更多!2022-11-23 09:36

對國產藥的評價需要回歸客觀公正的環境。
撰文丨周葉斌(美國阿拉巴馬大學伯明翰分校博士)
自11月11日國務院公佈了優化疫情防控的“二十條”之後,許多人開始個人儲備藥物,以應對防疫政策寬鬆後可能面臨的感染風險。11月18日,網上有藥店公開銷售國產抗新冠口服藥阿茲夫定片。然而,不到24小時,阿茲夫定片已緊急全網下架,廠家稱阿茲夫定是處方藥,不適合在家自行服用。這一“上網下網”風波,引起了大家的關注。
阿茲夫定(Azvudine,FNC)是河南真實生物科技有限公司(簡稱“真實生物”)研製、國內首款獲批的治療新冠病毒肺炎的小分子口服創新藥。2021年7月20日,阿茲夫定以抗艾滋病藥物通過中國國家藥監局附條件批准,老藥新用,又於今年7月25日獲藥監局附條件批准上市,用於治療新型冠狀病毒適應症,並被納入《新型冠狀病毒肺炎診療方案(第九版)》。阿茲夫定獲批治療新冠近四個月後忽然可以公開網購,又忽然下架,引發了網上“買到了能不能吃”的焦慮,這提醒着我們去關注、瞭解這款國產新藥的相關信息。
情況不容樂觀。
1 仍未發表的數據
阿茲夫定作為抗新冠口服藥獲批是通過緊急使用授權,從未公佈過完整的臨牀試驗數據。當然,這並不能阻止藥企宣稱數據已上報,正在整理,準備投到國際期刊發表。不過這些話是今年7月下旬獲批時説的,如今已然歲末,阿茲夫定要在國際期刊公佈的臨牀試驗數據在哪裏?
其實投稿國際期刊與公示數據沒有任何矛盾,研究人員完全可以將試驗結果以預印版形式先公開。自新冠疫情暴發以來,我們看到無數重要的新冠研究先以預印版論文公佈,再發表於期刊,生命科學各大頂級期刊無不接受這一操作流程。然而,三個多月來,除了打算建一年幾十億劑的生產線外,阿茲夫定未曾公開過任何數據。
我們能找到的也僅有8月相關藥企申請港股IPO的材料中極為有限的數據[1]。

就是這些IPO文件裏極為簡略的內容,仍然讓阿茲夫定看上去問題多多。本文分析如下。
2 試驗人數問題
在IPO文件裏提到阿茲夫定有三個臨牀試驗,分別在中國、俄羅斯與巴西進行,中國的已經完成,俄羅斯的也達到了設計的招募人數,而巴西的只完成了招募人數的一半[1]。
中國的試驗從2020年6月做到了2022年3月,計劃招募342人,實際招募348人,標準是輕症與普通型新冠。俄羅斯的試驗從2021年6月開始,計劃招募314人,已招募314人,標準是中症。巴西的試驗與俄羅斯一樣,在2021年6月開始,標準是中症,計劃招募342人,但只完成招募180人。
這些試驗的主要終點不一,中國的是載毒量下降,俄羅斯與巴西是症狀緩解。但無論什麼終點,計劃招募人數如此之少都是非比尋常的。
有人可能會説,管它招募多少人,最後有效性指標——臨牀試驗終點——能做出統計意義上的區別不就行了?可是要在非常少的招募人數下做出顯著差異,意味着藥效要非常好,這樣用藥組與安慰劑組才能拉開足夠的差距。但三期臨牀試驗是前瞻性研究,試驗完成前(包括設計試驗的時候),沒人知道藥效有多高。因此試驗人數的確定,一般是在“希望能有多大概率(統計檢驗功效)確認至少多高的有效性”這一基礎上去推算的。
比如新冠疫苗的臨牀試驗,假設希望能有90%的把握確認一個50%有效性的疫苗,可以回推需要多少病例,再根據一些感染率假設,推算應招募多少人以及試驗需要做多久。
新冠藥物在試驗設計階段,認為只要招募三百多人就能確定有效性,幾乎是匪夷所思的。
我們可以參考獲得FDA批准的輝瑞與默克兩家的口服藥。在輕到中症的高危人羣三期臨牀,輝瑞計劃招募約3000人,希望有1700人有數據做主要分析[2],默克則計劃招募1550人[3]。這些都是基於“希望有足夠的統計檢驗功效去檢測50%降低重症風險”而演算出來的需要的樣本量大小。最後兩家實際分析的數據量分別有2200多人與1400多人。
即使説這兩個藥檢測的降低重症風險與阿茲夫定的臨牀試驗終點不同,那參考輝瑞口服藥在低危人羣的EPIC-SR試驗——這裏主要終點是症狀持續改善——也招募了1440人[4]。
國內新冠單抗藥Brii-196/198,參與NIH的ACTIV-2試驗,確認有效性是用藥組418人,安慰劑組419人[5]。
為什麼阿茲夫定的研發方認為300多人的試驗就能驗證藥物有效性?同行都是計劃招募一兩千人來明確有效性的時候,有人卻不斷設計300多人的臨牀試驗,這是需要警惕的。
3 魔鬼在細節
阿茲夫定藥企的IPO文書中,中國與俄羅斯兩項臨牀試驗的陽性結果有很多細節值得關注,以下僅舉幾例。
中國的三期臨牀試驗中,主要終點是受試者服藥後第7與14天時的載毒量。可是在描述有效性時,文件中卻加了一個前提——基線載毒量高於3^10,在這些高載毒量受試者中,第3、5、7天用藥組載毒量下降比安慰劑組更多[1]:

那麼問題來了,這載毒量高的受試者是多少人呢?這一標準是事先確定的,還是事後加入?如果是事後加入,是否存在偏倚(bias)呢?而且即使是在這不知道多少人的高載毒量組裏,載毒量變化達到顯著差異的只有第5天。
根據這些描述可以推斷試驗的主要終點——受試者第7與14天的載毒量,用藥組與安慰劑組沒有顯著差異,也就是説該試驗沒有達到主要終點。
另外,IPO文件裏還提到所有次級終點均未顯示顯著差異。那麼次級終點裏有什麼呢?除了吸氧比例、肺炎變化等症狀,還有核酸檢測轉陰時間與速率。這與阿茲夫定獲批時的新聞稿裏“5天核酸轉陰”可是直接矛盾的。姑且不説新聞稿是否準確,核酸檢測結果與載毒量是有一定對應關係的,如果核酸轉陰時間、速率在用藥組都無改善,所謂基線高載毒量組第5天載毒量下降更多,這一陽性結果能有多靠譜呢?
再來看俄羅斯的試驗,這個試驗也是“阿茲夫定有助症狀緩解”的説法來源[1]:
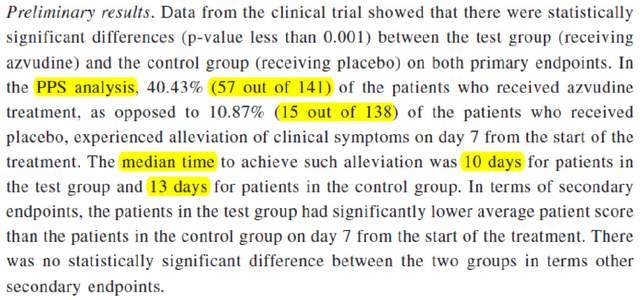
根據藥企的説法,第7天時,用藥組40.43%的人症狀有改善,而安慰劑組只有10.87%,差異非常大。可這是PPS分析,是去掉一些隨機入組但不符合試驗流程的受試者之後所做的分析。實際這個試驗兩組都已經招募了157人,但PPS分析時分別只有141與138人,其它的人是因為什麼原因被去除?如果沒被去除,結果是如何呢?
一般來説,PPS展示的是理想情況下藥物的有效性,因為各種不符合試驗理想流程的人都可以剔除。而ITT(意願治療分析)與FAS(全分析集)更貼近最初隨機入組的人羣,也更嚴格。要知道三期臨牀試驗能作為檢驗新藥有效性的金標準,很大一個原因是受試者分配入組是隨機的,而這個隨機是在招募時確定的。ITT就是真實反映了這個隨機狀態,而PPS涉及剔除掉任何“不符合試驗流程的受試者”,在保存試驗的隨機性上不如ITT或FAS(ITT中去掉完全沒用藥等極特殊情況)。
但是,無論是中國試驗還是俄羅斯試驗,IPO文件都未提供ITT或FAS的分析結果。中國試驗裏提到了FAS人數,可給出具體數據的卻只是高載毒量組這樣一個亞組分析,對“隨機”背離更遠了。
當一個藥企只提供亞組分析、PPS分析這類更寬鬆、潛在偏倚風險更大的結果,而沒有ITT、FAS數據,甚至連亞組與PPS具體標準都沒有時,我們必須要高度警覺。
此外,在俄羅斯試驗裏,第7天用藥組(下圖藍線)符合症狀緩解標準的已經達到40%,可中位症狀緩解時間卻是10天,也就是説第10天用藥組符合緩解標準的是50%。前7天與8-10這三天的緩解速率差異不小。安慰劑組(下圖橙色)則反了過來,第7天——也就是第一週還只有10%的人緩解,再過一週(第13天)卻有一半人緩解。走勢如下:
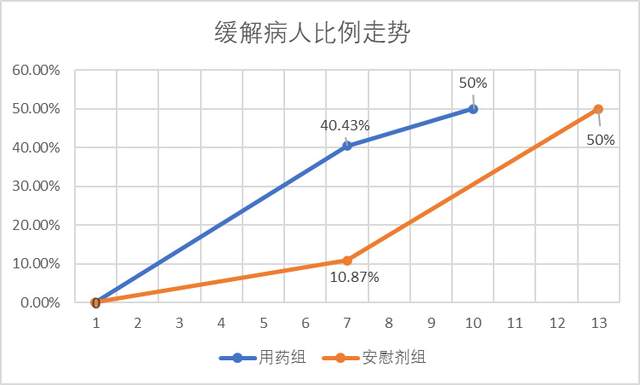
作者根據IPO數據繪製
緩解病人佔比的速率如此急轉也是較為罕見的情況,需要更詳細的數據公開,才能搞清楚其中的原因。但IPO提供的數據不夠詳實,無法對此做出任何解釋。
4 老藥就安全嗎?
IPO材料的信息透明度低,令阿茲夫定的有效性顯得極為可疑。不過,在關注有效性問題之外,我們更需要關注藥物的安全性問題。
文章開頭提到,阿茲夫定在2021年7月獲批用於HIV治療。有人説,阿茲夫定是老藥了,它的安全性是有保證的。但要知道,阿茲夫定作為抗HIV藥物,獲批依據是一個事先未設定標準的二期臨牀試驗的事後非劣性分析[6]。
在已經有大量高效HIV抗病毒藥上市的情況下,為什麼阿茲夫定能獲得如此“優待”,可以附條件優先審批?在沒有大規模三期臨牀試驗數據的情況下,只有295人使用過至少一劑治療就被允許上市[6]?
阿茲夫定最初作為抗HIV藥物時,對標的是拉米夫定(Lamivudine,3TC)。去搜阿茲夫定國內的報道,到處是“比拉米夫定劑量低”“不受拉米夫定耐藥性影響”的溢美之詞。
但是這些一邊倒的正面報道並沒有告訴我們,拉米夫定的遺傳毒性實驗結果[10]除了一個細胞實驗結果不明確,在體內體外均為陰性,而阿茲夫定則在體內體外均為陽性(下文會講到);拉米夫定的致癌性實驗結果為陰性,而阿茲夫定根本就缺乏致癌性實驗數據,還得補上。

拉米夫定的遺傳毒性實驗結果[10]
至於耐藥性,阿茲夫定的HIV臨牀試驗裏也出現了耐藥性突變[6]。不受拉米夫定的耐藥性突變影響,不代表HIV病毒不會對阿茲夫定產生耐藥性,只不過發生不同的耐藥突變而已。
當我們給中國的HIV感染者使用阿茲夫定而不是拉米夫定的時候,他(她)們知不知道這些差異,還是隻聽説阿茲夫定比拉米夫定更好?他(她)們最基本的知情權有沒有被尊重?
5 遺傳毒性
前文提到,阿茲夫定獲批抗HIV藥物根據的是一個臨牀二期非劣性試驗結果。也多虧一年前的這個批准,我們能找到當時藥品監管機構的技術評審報告[6]。
該報告提到,阿茲夫定在遺傳毒性實驗裏“Ames 試驗、CHL 染色體畸變試驗和體內小鼠微核試驗結果均為陽性”:

遺傳毒性(genotoxicity)是某個化學物質導致細胞內遺傳物質發生突變的能力。對於一個藥物,我們需要評估其遺傳毒性,因為萬一它能導致人體細胞的基因組發生突變,那就有潛在的致癌性、致畸性等嚴重安全問題。
遺傳毒性不是一刀切的風險。這體現在兩個層面。評估遺傳毒性的方法有很多,比如用實驗室培養的細胞,看看藥品有沒有在這些培養的細胞裏導致突變,也可以在動物實驗裏觀察有沒有突變發生;而同一個藥物用不同實驗,做出來的結果又可能不一樣。第一個層面就是説我們只能綜合考慮,給出一個風險上的評價。
從不同檢測方法的結果來看,阿茲夫定的評審報告包括了三種常用的遺傳毒性評估方法:①Ames是用細菌分析致突變性,②CHL染色體畸形是用小鼠細胞觀察,③體內小鼠微核試驗則是在動物身上直接研究。三者與人體的相關性也有一定的遞進關係,細菌是原核生物,與人體細胞相差甚遠,小鼠細胞就接近很多,但體外培養細胞與真實的藥物使用還是有很大差異,到小鼠體內試驗,基本是遺傳毒性在實驗階段能與人最接近的情況了。
這三項評估全部是陽性,也就是三種試驗都能觀察到阿茲夫定的致基因組突變現象,遺傳毒性可説是不容忽視。
遺傳毒性還要在另一個層面考慮,那就是藥品的風險收益。如果是不治之症,那麼對遺傳毒性的風險耐受度會更高,也就是對藥物的毒性標準會更寬容。畢竟,假如沒有其它替代藥物,那也不得不寬容。
到阿茲夫定的新冠治療上,遺傳毒性無論在哪個層面好像都不該寬容。參考同樣遇到潛在致突變風險的默克口服藥莫那匹韋(molnupiravir):在FDA上市審核時,遺傳毒性成了這個藥重點討論的話題(詳見《全球首款抗新冠病毒口服藥莫那匹韋,是天使,還是魔鬼?》)。實際上,在多種實驗裏,莫那匹韋只有一個Ames實驗陽性[7],動物體內實驗都是陰性(見下圖)。即便如此,FDA仍然對莫那匹韋做出了極為嚴格的限制——孕婦與未成年人直接排除在外,剩下的也必須在其它藥物——包括paxlovid和單克隆抗體——都不能用的情況下使用。
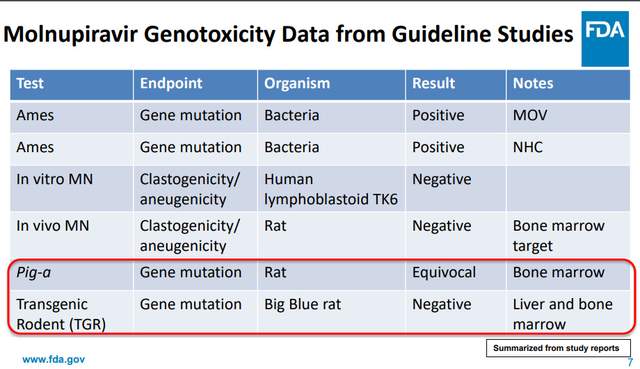
莫那匹韋是全球首款抗新冠口服藥。儘管如此,在其接受審核時,FDA多位專家提出,一旦有其它更好的藥物上市,就需要考慮撤銷莫那匹韋。此後,paxlovid橫空出世(詳見《無懼變異:輝瑞新藥Paxlovid或將破除新冠陰影》),莫那匹韋的使用範圍的確變得非常狹窄了。
現在阿茲夫定的遺傳毒性風險比莫那匹韋明確得多,藥物療效證據卻更少。莫那匹韋當時是一枝獨秀,且的確可以有效降低重症死亡風險,但現在市面上已經有多款其它抗病毒藥與單克隆抗體,阿茲夫定的上市還有必要嗎?
6 生殖毒性
阿茲夫定還存在明顯的生殖毒性風險。大鼠與兔兩種常用生殖毒性實驗動物中,有多個結果陽性[6]。
比如測試大鼠生育毒性:在交配前後給大鼠用藥,無論是給雌性還是雄性大鼠喂藥,高劑量時都會導致生育力下降:

阿茲夫定對胚胎髮育也有潛在風險。在大鼠妊娠早期給藥,會導致胚胎丟失率上升,5mg/kg的高劑量還會影響胚胎骨骼發育。
大鼠妊娠後期給藥則對母體與子代均有毒性,子代影響包括了一些生殖系統與神經系統影響:

這些結果在兔子的胚胎髮育毒性實驗裏也可以見到,包括上述生殖系統影響、胚胎存活降低以及胚胎骨骼發育等問題。
一些人可能會説這些生殖毒性實驗中所用劑量都比較高,比如大鼠妊娠晚期對母體有毒性的最低劑量(NOAEL)是0.5mg/kg,致死劑量是5mg/kg,而阿茲夫定治療新冠的人體劑量才5mg,按體重算比這些實驗劑量小很多。
但是,動物與人體在代謝水平上有差異,比如實驗用的大鼠、兔子,體型比人小很多,代謝快很多。考慮藥物劑量時需要做人體等效劑量的換算,比如,大鼠的劑量乘上0.162才是人體對應的劑量。這樣算下來,大鼠妊娠晚期的最低有毒性劑量0.5mg/kg,對應人體是0.081mg/kg,按標準體重60千克算,人體劑量是4.86毫克,比治療新冠用的5毫克還低。喜提零安全空間。
就算是雄性大鼠生育毒性的最低劑量高一點,5mg/kg,換算下來對應60千克的人,也就48.1毫克,跟治療新冠的劑量比,不到10倍的安全空間。如果用藥男性身形瘦小,只能説多保重了。
更重要的是,遺傳毒性、生殖毒性自有其特殊性,不是所謂的“不良反應”,臨牀試驗很難明確(更何況阿茲夫定HIV的臨牀試驗是個二期非劣性試驗,到目前為止所有HIV臨牀試驗加在一起,用過阿茲夫定的人應該在300個以下)。臨牀試驗裏沒觀察到,就説沒有,是很不嚴謹的。在藥物研發過程中,正確的做法是參考動物毒性試驗,一旦有,就認為是有潛在風險,就要考慮這類風險是否可接受。並不是説有毒性的劑量比較高,就要用較高的暴露劑量來探索潛在風險。
我們可以參考莫那匹韋的做法[2]:
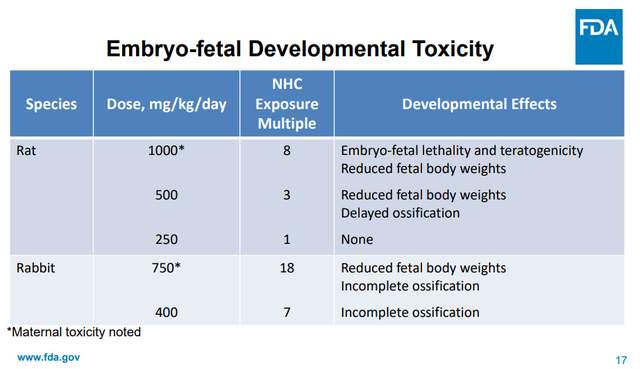
莫那匹韋的使用劑量是800mg,一天兩次,但生殖毒性實驗仍採用遠超這一水平的劑量檢測(在動物實驗中需要更高劑量去觀察,幫助明確這類風險)。莫那匹韋一樣觀察到了類似的胚胎髮育毒性,特別是骨骼發育影響(倒沒有阿茲夫定觀察到的對母體毒性)。需要注意的是,因為有這些毒性,莫那匹韋被絕對排除在孕婦與未成年人中使用,還被摁死在了其它治療藥物均不可用情況下的最末位選擇。
阿茲夫定的限制在哪裏?它的遺傳毒性類似於莫那匹韋,有效性卻不如後者明確,這個藥物在批准時是怎麼考量的?
綜上,阿茲夫定的安全性風險不是説我們一般説的常見不良反應或罕見不良反應,而是較為罕見的毒性問題。我們可以參考“吸煙致癌“——是否是每個抽煙的人都會得癌症?是否有一個明確的抽多少就會得癌症的標準?答案是沒有。可是因為煙草致癌的風險是存在的,就需要把這個風險納入管控的考量。藥物的生殖毒性與遺傳毒性類似,因為風險是存在的,就要納入“這藥能不能用”的權衡之中。
7 關鍵是總體風險
阿茲夫定針對HIV治療的技術評審報告中還提到,致癌性實驗尚未完成,需要補充[6]。考慮到這個藥已經表現出非常明確的致突變性,也就是遺傳毒性,致癌性的風險應該會是比較高的——癌變背後本身就是基因突變。
其實,更重要的是綜合各項數據分析阿茲夫定的風險。
阿茲夫定(Azvudine,FNC)是核苷類逆轉錄酶抑制劑,化學結構上是核苷類似物,與胞嘧啶類似(核酸序列ATCG中的C)。另一個HIV常用藥拉米夫定(Lamivudine,3TC)也是如此[8]:
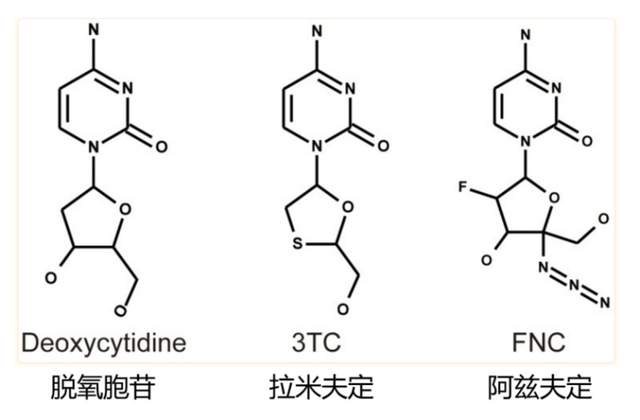
核苷類逆轉錄酶抑制劑是HIV藥物裏的一大類,它的結構與HIV逆轉錄複製基因組所需的原料——脱氧核糖核苷酸——類似,它與逆轉錄酶結合後可以阻斷病毒複製。
但這樣的機理必須關注遺傳毒性與生殖毒性問題。因為萬一結合的不是病毒的逆轉錄酶(對於新冠病毒來説就是RNA複製酶),而是人體DNA複製酶,影響了人體DNA的複製,那就有潛在的致突變風險。
不同的核苷類逆轉錄酶抑制劑的致突變風險不同。最早一個HIV藥物齊多夫定(AZT)是致突變性最高的,很多後來者要安全得多。但每一個藥物都需要獨立分析。
對於阿茲夫定來説,目前的證據都表明它的遺傳毒性、生殖毒性不可忽視。實際上還有其它研究指向阿茲夫定的這類風險。之前,有論文顯示阿茲夫定可以抑制腫瘤細胞複製[9],抑制的機理是阻斷了細胞分裂週期中DNA複製的那一階段。既然腫瘤細胞一樣是人的細胞,那麼這意味着阿茲夫定針對的並不只是病毒的逆轉錄酶或RNA複製酶,而是同樣會針對人體細胞的DNA複製酶。
更進一步來説,阿茲夫定抑制癌細胞複製的濃度並不高(微摩級別)——小鼠腫瘤模型的體內實驗也在比較低的劑量(0.5mg/kg,對應人體劑量0.0405mg/kg,標準體重劑量2.43毫克,新冠治療劑量的一半不到)就看到對人源腫瘤有抑制。這預示阿茲夫定影響人體DNA複製的效率並不低,更加證明了它有切實的遺傳毒性。
作為一個新冠治療藥物,阿茲夫定的用藥週期相對較短,這可能是對其遺傳毒性、生殖毒性問題唯一的安慰。但問題是,我們需要綜合考慮疾病的危險性、藥物的有效性和安全性,以及其它備選藥物是否存在。
目前,新冠病毒感染者絕大多數都可以自愈,特別是大量接種疫苗之後人羣重症風險更低了。在這種情況下,口服藥的安全性標準應該是非常高的。阿茲夫定表現出來的“症狀緩解”是否還有實際意義?此外,現在國內已經有paxlovid與國產單克隆抗體藥,這些藥物有效性更明確,也沒有嚴重的安全隱患。
08
藥物安全不分國籍
從IPO文件來看,抗新冠藥阿茲夫定的第一個人體臨牀試驗最低劑量從1mg開始。根據動物毒性實驗,最大無毒性劑量(NOAEL)對應人體是3mg,為了受試者的安全,一般人體試驗起始劑量應該從NOAEL劑量的十分之一開始。如果有切實證據支持更高的起始劑量,那也不是不可以,可阿茲夫定有什麼理由例外呢?
一些人很喜歡強調國產的重要性。但基本的藥物有效性、安全性標準不應該因為藥物的“國籍”而有鬆動。甚至對一些國產藥應該更嚴才對,因為某些國產藥説到底只會在中國上市,只有中國人會吃。對這些藥的安全性有效性放鬆監管,誰來保護吃藥的人?
對國產藥的評價需要回歸客觀公正的環境,動輒扣國際領先、填補空白的帽子,是一種過度的“溺愛”,最後既害了國產藥,也害了中國人。
參考文獻
[1] https://www1.hkexnews.hk/app/sehk/2022/104646/documents/sehk22080402059.pdf
[2] https://www.nejm.org/doi/full/10.1056/NEJMoa2118542
[3] https://www.nejm.org/doi/full/10.1056/NEJMoa2116044
[4] https://clinicaltrials.gov/ct2/show/record/NCT05011513
[5] https://www.briibio.com/news-detail.php?id=354
[6] https://file.wuxuwang.com/zhuce/ssypfiles/e5d36909ee1240d2e86b0c3a15e0ac29%E4%B8%8A%E5%B8%82%E5%AE%A1%E8%AF%84%E6%8A%A5%E5%91%8A.pdf
[7] https://www.fda.gov/media/154473/download
[8] https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4140803/
[9] https://pubmed.ncbi.nlm.nih.gov/21219886/
[10] https://www.merck.com/docs/product/safety-data-sheets/hh-sds/Doravirine%20and%20Lamivudine%20and%20Tenofovir%20Disoproxil%20Fumarate%20Bilayer%20Formulation_HH_MX_EN.pdf