全球首例!他們給嬰兒編輯了基因_風聞
医学界-医学界官方账号-为你提供可靠、有价值的内容是我们的存在方式。昨天 22:28
這是基因療法首次在人類患者中實現定製化臨牀應用,有望為數百種罕見疾病帶來變革性的精準醫療方案。
**撰文 |**燕小六
責編丨汪 航
當地時間5月15日,一個國際研究團隊在《新英格蘭醫學雜誌》發表論文稱,他們成功為一名患有罕見遺傳病的嬰兒實施了定製化基因編輯治療。
這名9個月大的嬰兒患有一種無法有效代謝蛋白質的致命罕見病。研究人員利用CRISPR基因編輯技術開發出定製化療法,在嬰兒的肝細胞中成功糾正致病的特定基因突變。
從確診到接受治療,過程僅耗時6個月。截至今年4月,患兒已接受3劑次基因編輯治療,未發生嚴重不良反應。在他體內,因基因突變而受損的肝細胞被部分修復,血液中的有毒氨積聚也得以逆轉。
美國國立衞生研究院當天發表聲明稱,這是基因療法首次在人類患者中實現定製化臨牀應用,為將來開發針對其他罕見病的定製化基因療法奠定了基礎。
“它標誌着新的醫學時代來臨。成千上萬被傳統制藥市場忽視的罕見遺傳病患者,或有望通過定製基因療法而治癒。”《華盛頓郵報》評論稱。
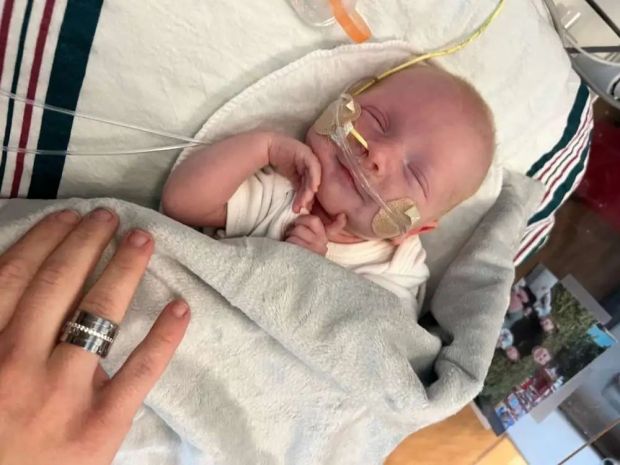
當地時間5月15日,《新英格蘭醫學雜誌》報告全球首例嬰兒期定製化基因編輯療法。圖為患兒KJ Muldoon近影。/圖源:Children’s Hospital of Philadelphia
首個定製化基因治療誕生背後
患兒名叫KJ Muldoon,患有氨甲酰磷酸合成酶1(CPS1)缺乏症,這是一種遺傳代謝病,迄今全球公開報告病例僅數百人。
攜帶相關基因突變者可能在任何年齡段發病,出現不同程度的高氨血癥、代謝性腦病及肝病。其中,新生兒期就發病的人,病情多進展迅速,病死率高,50%者無法活到成年。
2024年8月,KJ Muldoon出生的第二天,就出現了嗜睡、呼吸窘迫、肌肉僵硬等症狀。他的胳膊若被抬起,鬆開後就會顫抖着垂落到身體一側。血檢提示,他體內的氨含量達到危險水平。
一週不到,基因測序結果出來了。醫學團隊確認KJ Muldoon攜帶源自父母雙方的基因突變,診斷為CPS1缺乏症。
過往,針對該病的治療方案極有限,僅僅是嚴格控制蛋白質攝入;服用藥物、接受透析,去除體內不斷積聚的氨;以及根據病情、做肝移植。
5個月大時,KJ Muldoon被列入肝移植名單。據美國相關規定,患兒滿1歲或達到一定體重,才能做此類手術。
但KJ Muldoon很可能等不到,因為他的情況非常兇險,即使攝入微量蛋白質,也會導致血液中氨濃度激增。每經歷1小時高血氨狀態,大腦就會遭受不可逆損傷。
此時,美國費城兒童醫院團隊提出一種“創新思路”:可以利用鹼基編輯技術,精準地修復一個關鍵的基因缺陷,從而改善病症。
多年來,費城團隊一直在探索“定製化的基因編輯療法”。隨着研究深入,他們的研發流程不斷加速,從1-2年能設計出一套方案,縮短到只需幾個月就能完成,且在小鼠實驗中證明有效。
拿到KJ Muldoon的測序結果後,費城團隊迅速行動,開啓了一場生命大接力。
他們聯繫多名基因編輯療法專家,包括鹼基編輯技術發明者David Liu,長期倡導加速CRISPR藥物開發的加州大學伯克利分校科學家Fyodor Urnov等,組建起“CRISPR治癒聯盟”。
“聯盟”着手創建攜帶突變基因的細胞系、小鼠模型;不斷測試基因編輯器在肝細胞中的效用,試圖用最短時間、篩選出最優者……美國國立衞生研究院也參與其中,資助了一項針對非人類靈長目動物的毒理學實驗。
緊接着,不同地區、多家生物製藥公司陸續跟進,生產編輯技術和遞送系統等。一般情況下,由於經濟成本、倫理、法務風險等,這些流程通常不可能並行推進。
但這一次,所有人都為了孩子頂着巨大壓力開展研究,僅用6周時間,團隊就鎖定了最優研發方案。
直到此時,研究團隊才將定製化的基因編輯療法,對KJ Muldoon的父母全盤托出。“我們反覆權衡,仔細向醫護團隊諮詢肝移植和這種創新療法。我們祈禱、諮詢親友、收集信息,最終決定賭一把。”其父母回憶。
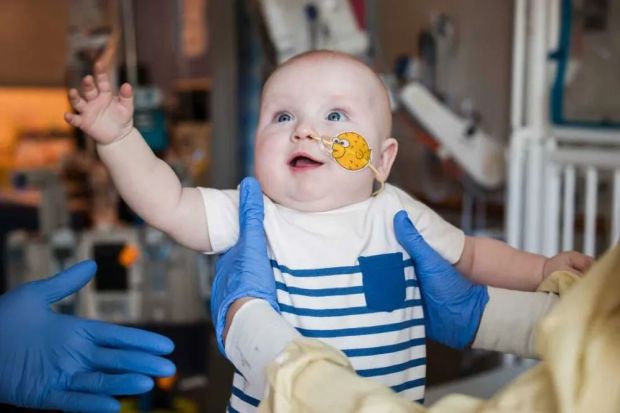
圖源:Children’s Hospital of Philadelphia
6個月定製“個性化治療方案”,起效了!
2024年11月的感恩節前夕,費城團隊與美國食品藥品監督管理局(FDA)首次舉行非公開會議,介紹前述定製療法。
費城團隊稱,雖然整個研發過程耗時較短,但該療法的完成度極高。在快速構建、攜帶目標基因片段的動物模型中,團隊驗證了它的安全性和有效性,已達到適用於人體的藥物生產階段。
今年情人節當天,費城團隊正式向FDA遞交臨牀申請。僅僅7天,團隊拿到FDA的緊急審查許可。這款定製藥物以KJ Muldoon之名,被命名為“kayjayguran abengcemeran”,簡稱k-abe。
2025年2月25日,通過靜脈輸注,患兒完成了第一劑次基因編輯治療。新一代鹼基編輯系統通過脂質納米顆粒(LNP)載體,遞送至他的肝細胞。在那裏,鹼基編輯器會被引導至變異基因處,直接修改單個鹼基,糾正致病的突變。
費城團隊兒科醫生、基因療法專家Rebecca Ahrens-Nicklas回憶,當天治療室內充滿興奮的喧鬧聲,“他(KJ Muldoon)全程酣睡。”
今年3、4月,KJ Muldoon又分別完成一次基因編輯治療。監測數據顯示,他的血氨濃度、谷氨醯胺水平、尿氨酸水平等,均向正常範圍回落。肝功能指標雖有輕度波動,但未見嚴重不良反應。神經系統也沒有出現進一步損害。
Rebecca Ahrens-Nicklas等人非常謹慎,不敢輕言“治癒”。因為要判斷基因編輯是否成功,必須做肝穿刺活檢,這對KJ Muldoon來説太過危險。
但可以肯定的是,大量跡象表明,患兒比治療前明顯好轉。服用較低劑量藥物後,他的日常蛋白質攝入恢復到同齡兒水平,健康狀況持續改善,體重穩步增長。他能正常坐起,像同齡兒一樣充滿活力。
費城團隊稱,KJ Muldoon有望在今年6月左右出院、和家人團聚。

醫療團隊和患兒合影。/圖源:Children’s Hospital of Philadelphia
鹼基編輯立大功
“在這麼短的時間內,成功開發出個性化的基因編輯療法,是醫學史重要的里程碑。我們由此踏入了一個嶄新的時代,一個曾經無法治癒的遺傳疾病也有望得到有效治療的時代。”基因編輯療法的開創者之一、諾貝爾獎得主Jennifer A. Doudna如此評論。
還有專家指出,鹼基編輯的問世,是促成前述“不可思議事件”的功臣。
鹼基編輯療法被稱為“下一代基因編輯技術”,它無需切斷DNA雙鏈,就能精準、永久性地編輯DNA中單個鹼基,校正致病性突變,在確保DNA完整性的同時,能一次性徹底實現疾病治癒,具有更高的安全性,編輯效率也更高。
目前,基因鹼基編輯已運用到T細胞白血病、高膽固醇血癥等治療探索中。2024年1月,廣西醫科大學第一附屬醫院發起臨牀試驗,於全球首次通過鹼基編輯療法,臨牀治癒一名重型β-地中海貧血症患者。
《新英格蘭醫學雜誌》針對前述費城團隊創新,發表隨附報告稱,這標誌着針對罕見病治療的基因編輯,從實驗室邁向臨牀,提供了全新思路,但仍需在安全性、可及性及倫理層面持續突破。
目前,針對KJ Muldoon的隨訪數據僅有7周,長期安全性、療效(如肝功能、神經發育)尚不明確。後續要長期追蹤其肝功能、血氨水平、神經發育及潛在副作用(如免疫反應、脱靶效應)。
美國斯坦福大學法學院生物倫理學家Henry Greely在回覆媒體的電子郵件中稱,此類研究需要考慮支付問題。據費城團隊表述,數十位合著者、聯邦資金、私人公司對於研究都有貢獻,不乏大額捐贈,無法計算出KJ Muldoon治療究竟花了多少錢。
Fyodor Urnov是加州大學伯克利分校創新基因組學研究所技術科學總監,參與了前述療法開發。他表示:“我們希望能開創先例。這將是基因編輯真正實現個體化治療的元年。”
(凌駿對本文亦有貢獻)
參考文獻:
1.Kiran Musunuru, et al. Patient-Specific In Vivo Gene Editing to Treat a Rare Genetic Disease. NEJM. May 15, 2025.
DOI:10.1056/NEJMoa2504747
2.董慧, 楊豔玲. 氨甲酰磷酸合成酶1缺乏症的診斷與治療研究進展. 中國實用兒科雜誌. 2022,Vol. 37, Issue (10): 766-770.
DOI:10.19538/j.ek2022100612